Nikolic Ana, Ricci Giulia, Sera Francesco, Bucci Elisabetta, Govi Monica, Mele Fabiano, Rossi Marta, Ruggiero Lucia, Vercelli Liliana, Ravaglia Sabrina, Brisca Giacomo, Fiorillo Chiara, Villa Luisa, Maggi Lorenzo, Cao Michelangelo, D'Amico Maria Chiara, Siciliano Gabriele, Antonini Giovanni, Santoro Lucio, Mongini Tiziana, Moggio Maurizio, Morandi Lucia, Pegoraro Elena, Angelini Corrado, Di Muzio Antonio, Rodolico Carmelo, Tomelleri Giuliano, Grazia D'Angelo Maria, Bruno Claudio, Berardinelli Angela, Tupler Rossella
Department of Science of Life, Institute of Biology, University of Modena and Reggio Emilia, Modena, Italy.
Department of Science of Life, Institute of Biology, University of Modena and Reggio Emilia, Modena, Italy Department of Clinical and Experimental Medicine, Neurological Clinic, University of Pisa, Pisa, Italy.
BMJ Open. 2016 Jan 5;6(1):e007798. doi: 10.1136/bmjopen-2015-007798.
Facioscapulohumeral muscular dystrophy type 1 (FSHD1) has been genetically linked to reduced numbers (≤ 8) of D4Z4 repeats at 4q35. Particularly severe FSHD cases, characterised by an infantile onset and presence of additional extra-muscular features, have been associated with the shortest D4Z4 reduced alleles with 1-3 repeats (1-3 DRA). We searched for signs of perinatal onset and evaluated disease outcome through the systematic collection of clinical and anamnestic records of de novo and familial index cases and their relatives, carrying 1-3 DRA.
Italy.
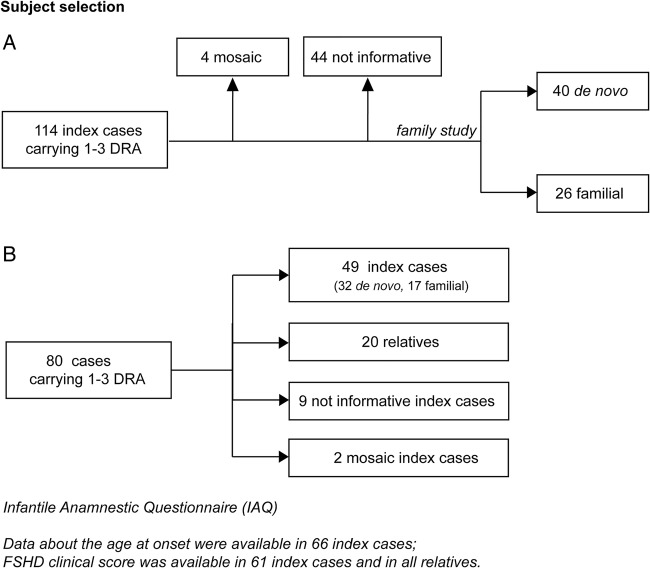
66 index cases and 33 relatives carrying 1-3 DRA.
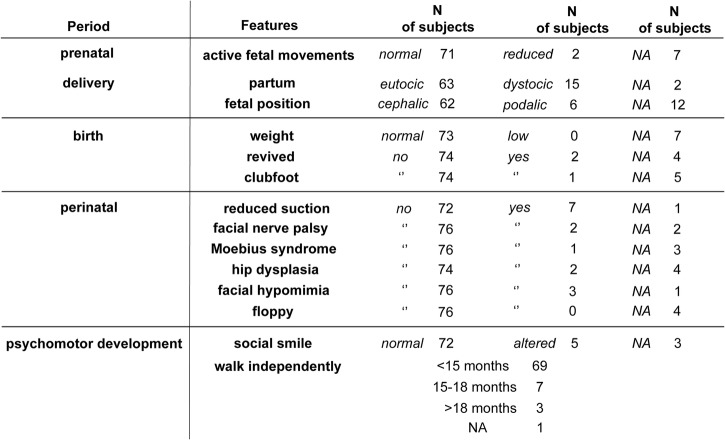
The clinical examination was performed using the standardised FSHD evaluation form with validated inter-rater reliability. To investigate the earliest signs of disease, we designed the Infantile Anamnestic Questionnaire (IAQ). Comparison of age at onset was performed using the non-parametric Wilcoxon rank-sum or Kruskal-Wallis test. Comparison of the FSHD score was performed using a general linear model and Wald test. Kaplan-Meier survival analysis was used to estimate the age-specific cumulative motor impairment risk.
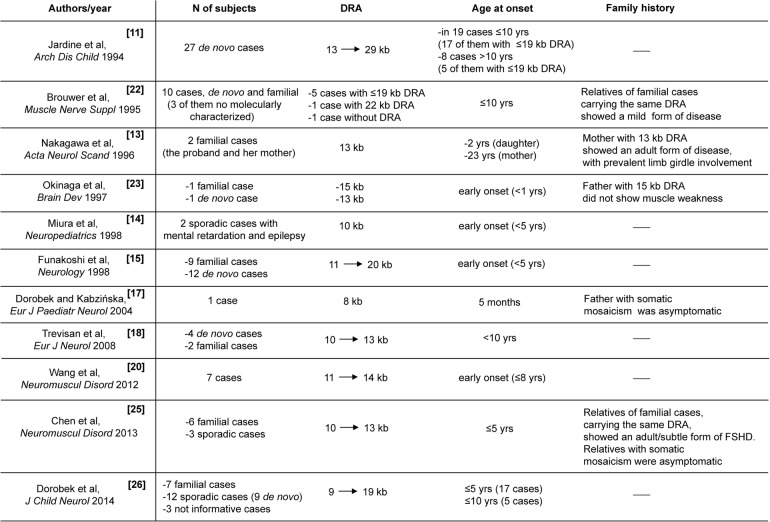
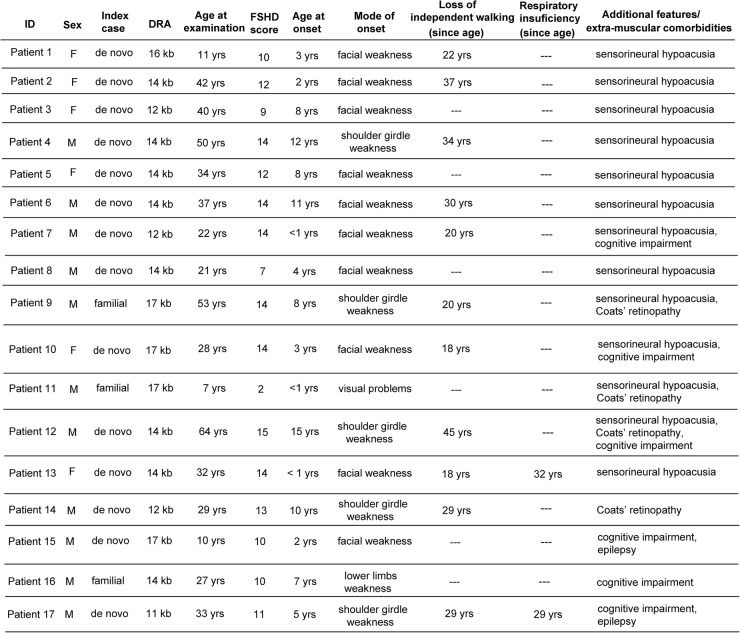
No patients had perinatal onset. Among index cases, 36 (54.5%) showed the first signs by 10 years of age. The large majority of patients with early disease onset (26 out of 36, 72.2%) were de novo; whereas the majority of patients with disease onset after 10 years of age were familial (16, 53.3%). Comparison of the disease severity outcome between index cases with age at onset before and over 10 years of age, failed to detect statistical significance (Wald test p value=0.064). Of 61 index cases, only 17 (27.9%) presented extra-muscular conditions. Relatives carrying 1-3 DRA showed a large clinical variability ranging from healthy subjects, to patients with severe motor impairment.
The size of the D4Z4 allele is not always predictive of severe clinical outcome. The high degree of clinical variability suggests that additional factors contribute to the phenotype complexity.
1型面肩肱型肌营养不良症(FSHD1)在基因上与4q35处D4Z4重复序列数量减少(≤8个)有关。以婴儿期发病和存在其他肌肉外特征为特征的特别严重的FSHD病例,与最短的具有1 - 3个重复序列的D4Z4减少等位基因(1 - 3 DRA)有关。我们通过系统收集携带1 - 3 DRA的新发和家族性索引病例及其亲属的临床和既往史记录,寻找围产期发病的迹象并评估疾病结局。
意大利。
66例索引病例和33名携带1 - 3 DRA的亲属。
使用具有经过验证的评分者间信度的标准化FSHD评估表进行临床检查。为了调查疾病的最早迹象,我们设计了婴儿既往史问卷(IAQ)。使用非参数Wilcoxon秩和检验或Kruskal - Wallis检验对发病年龄进行比较。使用一般线性模型和Wald检验对FSHD评分进行比较。采用Kaplan - Meier生存分析来估计特定年龄的累积运动障碍风险。
没有患者在围产期发病。在索引病例中,36例(54.5%)在10岁前出现了最初症状。大多数疾病早期发病的患者(36例中的26例,72.2%)是新发的;而大多数10岁后发病的患者是家族性的(16例,53.3%)。对发病年龄在10岁之前和之后的索引病例之间的疾病严重程度结局进行比较,未发现统计学意义(Wald检验p值 = 0.064)。在61例索引病例中,只有17例(27.9%)出现了肌肉外病症。携带1 - 3 DRA的亲属表现出很大的临床变异性,从健康受试者到严重运动障碍患者都有。
D4Z4等位基因的大小并不总是能预测严重的临床结局。高度的临床变异性表明其他因素导致了表型的复杂性。