Schilter Kala F, Reis Linda M, Sorokina Elena A, Semina Elena V
Department of Pediatrics and Children's Research InstituteMedical College of WisconsinMilwaukeeWisconsin53226; Department of Cell Biology, Neurobiology and AnatomyMedical College of WisconsinMilwaukeeWisconsin53226.
Department of Pediatrics and Children's Research Institute Medical College of Wisconsin Milwaukee Wisconsin 53226.
Mol Genet Genomic Med. 2015 Jun 2;3(6):490-9. doi: 10.1002/mgg3.159. eCollection 2015 Nov.
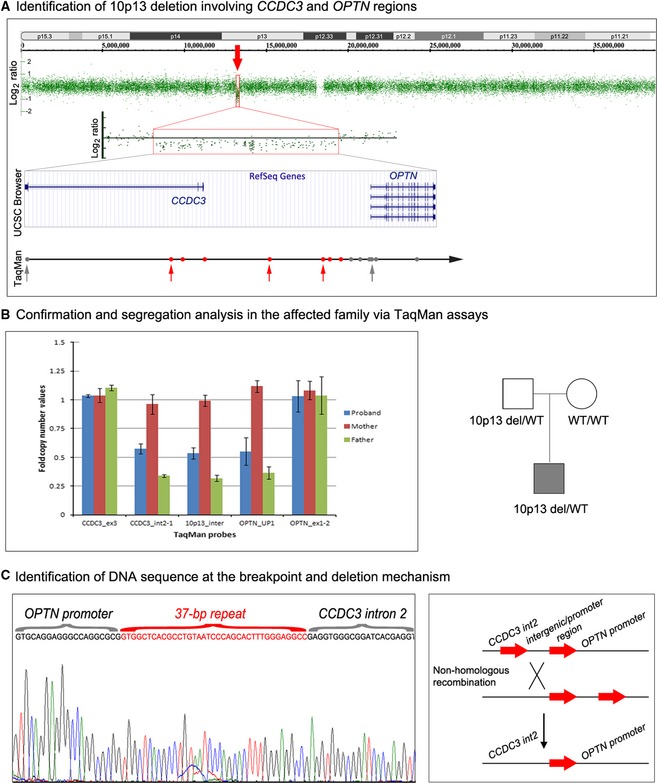
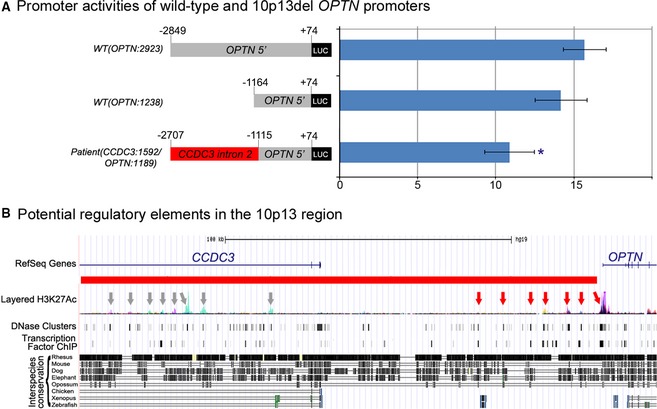
Genetic causes of ocular conditions remain largely unknown. To reveal the molecular basis for a congenital ocular phenotype associated with glaucoma we performed whole-exome sequencing (WES) and whole-genome copy number analyses of patient DNA. WES did not identify a causative variant. Copy number variation analysis identified a deletion of 10p13 in the patient and his unaffected father; the deletion breakpoint contained a single 37-bp sequence that is normally present in two distinct Alu repeats separated by ~181 kb. The deletion removed part of the upstream region of optineurin (OPTN) as well as the upstream sequence and two coding exons of coiled-coil domain containing 3 (CCDC3); analysis of the patient's second allele showed normal OPTN and CCDC3 sequences. Studies of zebrafish orthologs identified expression in the developing eye for both genes. OPTN is a known factor in dominant adult-onset glaucoma and Amyotrophic Lateral Sclerosis (ALS). The deletion eliminates 98 kb of the OPTN upstream sequence leaving only ~1 kb of the proximal promoter region. Comparison of transcriptional activation capability of the 3 kb normal and the rearranged del(10)(p13) OPTN promoter sequences demonstrated a statistically significant decrease for the deleted allele; sequence analysis of the entire deleted region identified multiple conserved elements with possible cis-regulatory activity. Additional screening of CCDC3 indicated that heterozygous loss-of-function alleles are unlikely to cause congenital ocular disease. In summary, we report the first regulatory region deletion involving OPTN, caused by Alu-mediated nonallelic homologous recombination and possibly contributing to the patient's ocular phenotype. In addition, our data indicate that Alu-mediated rearrangements of the OPTN upstream region may represent a new source of affected alleles in human conditions. Evaluation of the upstream OPTN sequences in additional ocular and ALS patients may help to determine the role of this region, if any, in human disease.
眼部疾病的遗传病因在很大程度上仍然未知。为了揭示与青光眼相关的先天性眼部表型的分子基础,我们对患者的DNA进行了全外显子组测序(WES)和全基因组拷贝数分析。WES未发现致病变异。拷贝数变异分析在患者及其未受影响的父亲中发现了10p13缺失;缺失断点包含一个37bp的序列,该序列通常存在于两个相距约181kb的不同Alu重复序列中。该缺失去除了视紫质(OPTN)上游区域的一部分以及包含卷曲螺旋结构域3(CCDC3)的上游序列和两个编码外显子;对患者第二个等位基因的分析显示OPTN和CCDC3序列正常。对斑马鱼直系同源基因的研究确定了这两个基因在发育中的眼睛中的表达。OPTN是成人显性青光眼和肌萎缩侧索硬化症(ALS)的已知因素。该缺失消除了OPTN上游序列的98kb,仅留下约1kb的近端启动子区域。对3kb正常和重排的del(10)(p13) OPTN启动子序列的转录激活能力进行比较,结果显示缺失等位基因有统计学意义的下降;对整个缺失区域的序列分析确定了多个具有可能顺式调节活性的保守元件。对CCDC3的进一步筛查表明,杂合功能丧失等位基因不太可能导致先天性眼部疾病。总之,我们报告了首例由Alu介导的非等位基因同源重组导致的涉及OPTN的调控区域缺失,这可能导致了患者的眼部表型。此外,我们的数据表明,Alu介导的OPTN上游区域重排可能是人类疾病中受影响等位基因的新来源。对更多眼部和ALS患者的OPTN上游序列进行评估,可能有助于确定该区域在人类疾病中的作用(如果有)。