Meyer Austin G, Spielman Stephanie J, Bedford Trevor, Wilke Claus O
Department of Integrative Biology, Institute for Cellular and Molecular Biology, and Center for Computational Biology and Bioinformatics. The University of Texas at Austin, Austin, TX; School of Medicine, Texas Tech University Health Sciences Center, Lubbock, TX.
Department of Integrative Biology, Institute for Cellular and Molecular Biology, and Center for Computational Biology and Bioinformatics. The University of Texas at Austin, Austin, TX.
Virus Evol. 2015 Jan;1(1). doi: 10.1093/ve/vev006. Epub 2015 Jan 1.
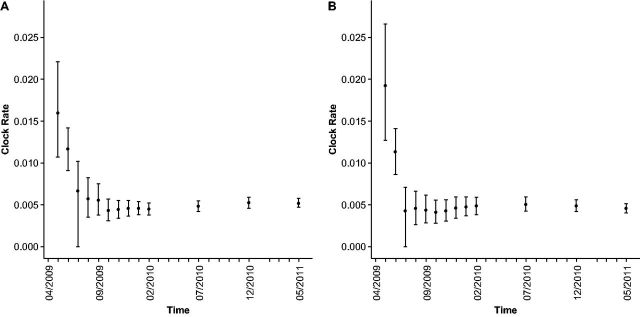
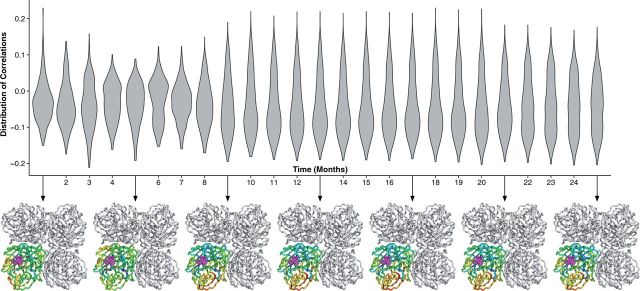
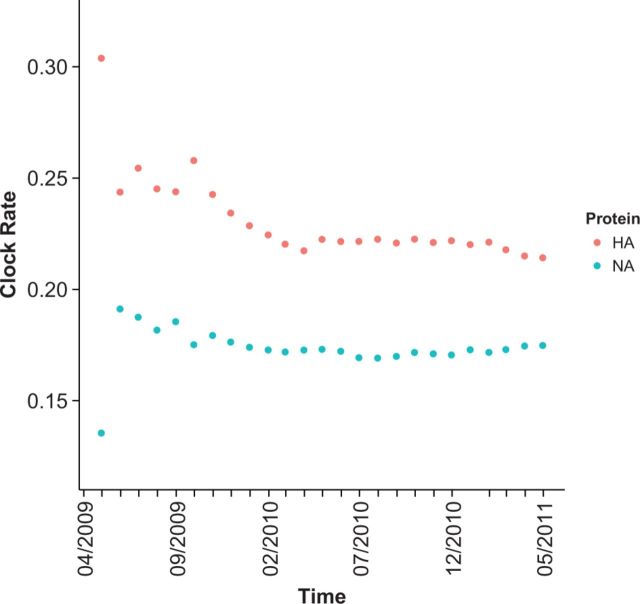
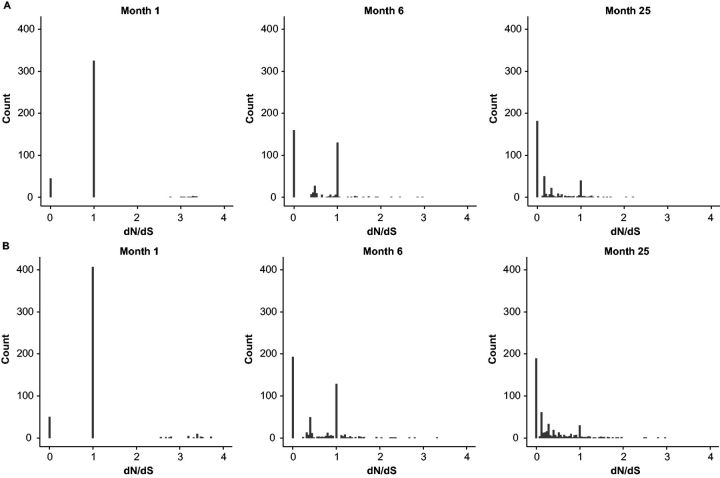
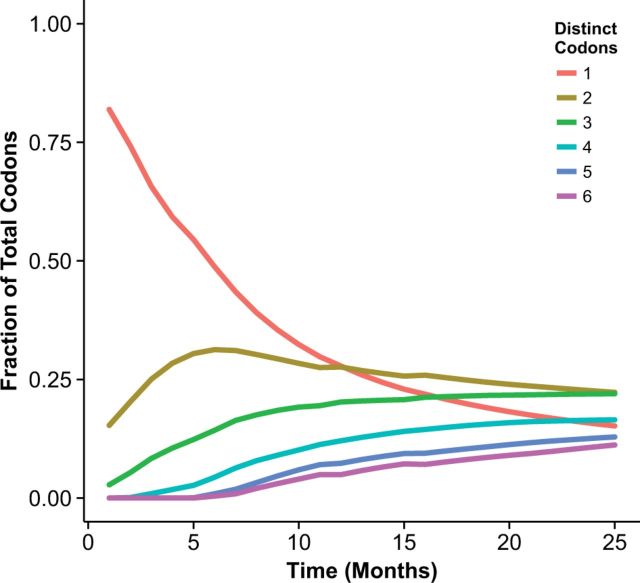
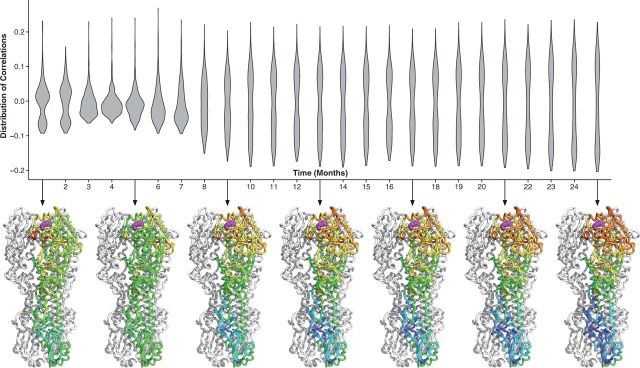
With the expansion of DNA sequencing technology, quantifying evolution in emerging viral outbreaks has become an important tool for scientists and public health officials. Although it is known that the degree of sequence divergence significantly affects the calculation of evolutionary metrics in viral outbreaks, the extent and duration of this effect during an actual outbreak remains unclear. We have analyzed how limited divergence time during an early viral outbreak affects the accuracy of molecular evolutionary metrics. Using sequence data from the first 25 months of the 2009 pandemic H1N1 (pH1N1) outbreak, we calculated each of three different standard evolutionary metrics-molecular clock rate (i.e., evolutionary rate), whole gene , and site-wise -for hemagglutinin and neuraminidase, using increasingly longer time windows, from 1 month to 25 months. For the molecular clock rate, we found that at least three to four months of temporal divergence from the start of sampling was required to make precise estimates that also agreed with long-term values. For whole gene , we found that at least two months of data were required to generate precise estimates, but six to nine months were required for estimates to approach their long term values. For site-wise estimates, we found that at least six months of sampling divergence was required before the majority of sites had at least one mutation and were thus evolutionarily informative. Furthermore, eight months of sampling divergence was required before the site-wise estimates appropriately reflected the distribution of values expected from known protein-structure-based evolutionary pressure in influenza. In summary, we found that evolutionary metrics calculated from gene sequence data in early outbreaks should be expected to deviate from their long-term estimates for at least several months after the initial emergence and sequencing of the virus.
随着DNA测序技术的发展,在新出现的病毒爆发中量化进化已成为科学家和公共卫生官员的一项重要工具。尽管已知序列差异程度会显著影响病毒爆发中进化指标的计算,但在实际爆发期间这种影响的程度和持续时间仍不清楚。我们分析了早期病毒爆发期间有限的分化时间如何影响分子进化指标的准确性。利用2009年甲型H1N1流感(pH1N1)大流行爆发头25个月的序列数据,我们使用从1个月到25个月越来越长的时间窗口,计算了血凝素和神经氨酸酶的三种不同标准进化指标——分子钟速率(即进化速率)、全基因和位点特异性指标。对于分子钟速率,我们发现从采样开始至少需要三到四个月的时间差异才能做出精确估计,且这些估计值也与长期值相符。对于全基因指标,我们发现至少需要两个月的数据才能产生精确估计,但估计值接近其长期值则需要六到九个月。对于位点特异性估计,我们发现至少需要六个月的采样差异,大多数位点才会至少有一个突变,从而具有进化信息。此外,在位点特异性估计能够恰当地反映基于已知蛋白质结构的流感进化压力所预期的值的分布之前,需要八个月的采样差异。总之,我们发现,在病毒最初出现并测序后的至少几个月内,早期爆发中从基因序列数据计算出的进化指标应会偏离其长期估计值。