Fu Yong-Bi, Peterson Gregory W, Dong Yibo
Plant Gene Resources of Canada, Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada, Saskatoon, Saskatchewan S7N 0X2, Canada
Plant Gene Resources of Canada, Saskatoon Research and Development Centre, Agriculture and Agri-Food Canada, Saskatoon, Saskatchewan S7N 0X2, Canada.
G3 (Bethesda). 2016 Apr 7;6(4):845-56. doi: 10.1534/g3.115.025775.
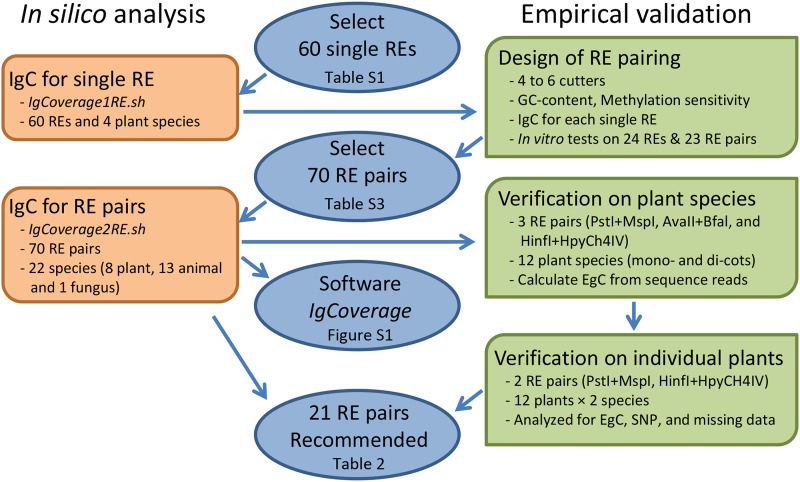
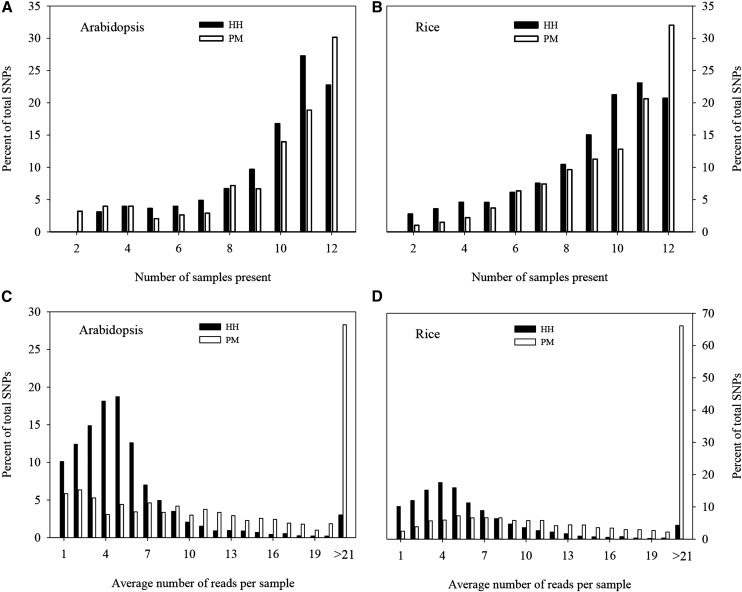
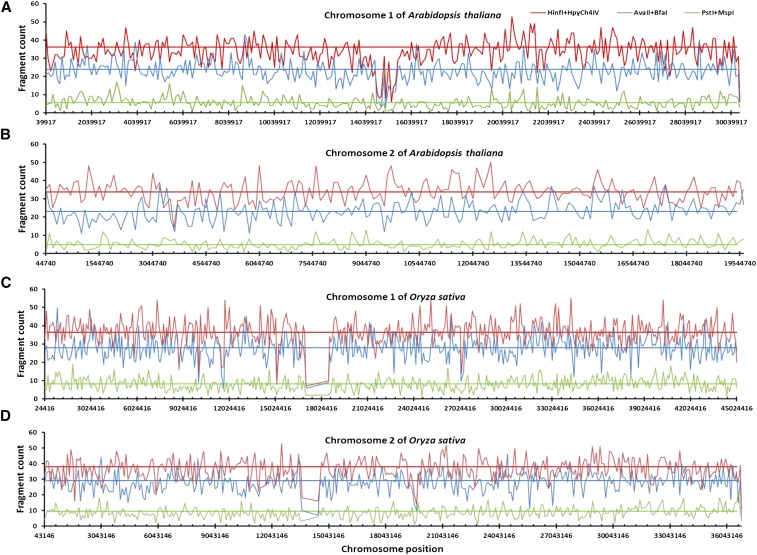
Genotyping-by-sequencing (GBS) has emerged as a useful genomic approach for exploring genome-wide genetic variation. However, GBS commonly samples a genome unevenly and can generate a substantial amount of missing data. These technical features would limit the power of various GBS-based genetic and genomic analyses. Here we present software called IgCoverage for in silico evaluation of genomic coverage through GBS with an individual or pair of restriction enzymes on one sequenced genome, and report a new set of 21 restriction enzyme combinations that can be applied to enhance GBS applications. These enzyme combinations were developed through an application of IgCoverage on 22 plant, animal, and fungus species with sequenced genomes, and some of them were empirically evaluated with different runs of Illumina MiSeq sequencing in 12 plant species. The in silico analysis of 22 organisms revealed up to eight times more genome coverage for the new combinations consisted of pairing four- or five-cutter restriction enzymes than the commonly used enzyme combination PstI + MspI. The empirical evaluation of the new enzyme combination (HinfI + HpyCH4IV) in 12 plant species showed 1.7-6 times more genome coverage than PstI + MspI, and 2.3 times more genome coverage in dicots than monocots. Also, the SNP genotyping in 12 Arabidopsis and 12 rice plants revealed that HinfI + HpyCH4IV generated 7 and 1.3 times more SNPs (with 0-16.7% missing observations) than PstI + MspI, respectively. These findings demonstrate that these novel enzyme combinations can be utilized to increase genome sampling and improve SNP genotyping in various GBS applications.
基于测序的基因分型(GBS)已成为一种用于探索全基因组遗传变异的有用基因组方法。然而,GBS通常对基因组进行不均匀采样,并可能产生大量缺失数据。这些技术特征将限制各种基于GBS的遗传和基因组分析的效能。在此,我们展示了一款名为IgCoverage的软件,用于在计算机上评估通过GBS对一个测序基因组使用单个或一对限制性内切酶时的基因组覆盖情况,并报告了一组新的21种限制性内切酶组合,可用于增强GBS的应用。这些酶组合是通过将IgCoverage应用于22个具有测序基因组的植物、动物和真菌物种而开发的,其中一些在12个植物物种中通过不同批次的Illumina MiSeq测序进行了实证评估。对22种生物的计算机分析表明,由四切或五切限制性内切酶配对组成的新组合的基因组覆盖度比常用的酶组合PstI + MspI高出多达八倍。对12个植物物种中新型酶组合(HinfI + HpyCH4IV)的实证评估表明,其基因组覆盖度比PstI + MspI高1.7至6倍,在双子叶植物中的基因组覆盖度比单子叶植物高2.3倍。此外,对12株拟南芥和12株水稻的SNP基因分型显示,HinfI + HpyCH4IV分别比PstI + MspI产生多7倍和1.3倍的SNP(缺失观测值为0 - 16.7%)。这些发现表明,这些新型酶组合可用于增加基因组采样,并改善各种GBS应用中的SNP基因分型。