Martin Angela M, Maradei Silvia J, Velasco Harvy M
School of Medicine. Instituto de Genética, Universidad Nacional de Colombia, Bogota, Colombia.
Colomb Med (Cali). 2015 Dec 30;46(4):194-8.
Mutations of GDAP1 gene cause autosomal dominant and autosomal recessive Charcot-Marie-Tooth disease and more than 40 different mutations have been reported. The recessive Q163X mutation has been described in patients of Spanish ancestry, and a founder mutation in South American patients, originating in Spain has been demonstrated.
We describe physical and histological features, and the molecular impact of mutation Q163X in a Colombian family.
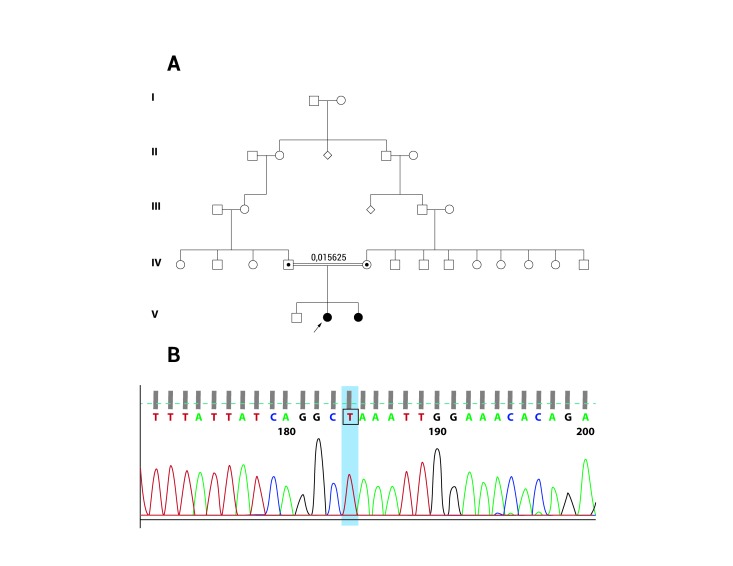
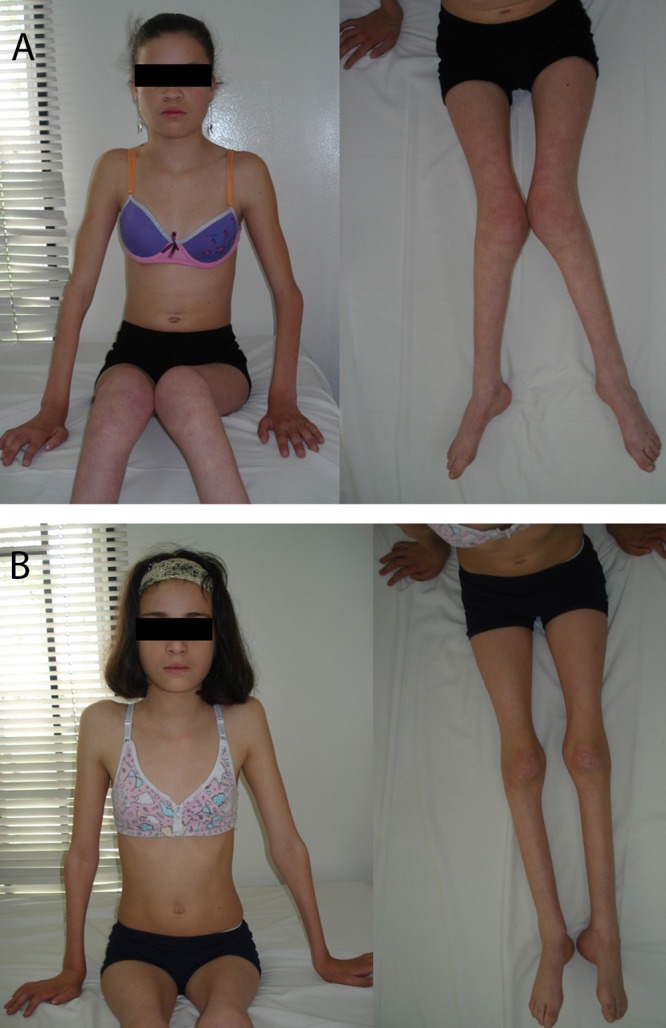
We report two female patients, daughters of consanguineous parents, with onset of symptoms within the first two years of life, developing severe functional impairment, without evidence of dysmorphic features, hoarseness or diaphragmatic paralysis. Electrophysiology tests showed a sensory and motor neuropathy with axonal pattern. Sequencing of GDAP1 gene was requested and the study identified a homozygous point mutation (c.487 C>T) in exon 4, resulting in a premature stop codon (p.Q163X). This result confirms the diagnosis of Charcot-Marie-Tooth disease, type 4A.
The patients were referred to Physical Medicine and Rehabilitation service, in order to be evaluated for ambulation assistance. They have been followed by Pulmonology service, for pulmonary function assessment and diaphragmatic paralysis evaluation. Genetic counseling was offered. The study of the genealogy of the patient, phenotypic features, and electrophysiological findings must be included as valuable tools in the clinical approach of the patient with Charcot-Marie-Tooth disease, in order to define a causative mutation. In patients of South American origin, the presence of GDAP1 gene mutations should be considered, especially the Q163X mutation, as the cause of CMT4A disease.
GDAP1基因突变可导致常染色体显性和常染色体隐性遗传性腓骨肌萎缩症,目前已报道了40多种不同的突变。隐性Q163X突变已在西班牙裔患者中被描述,并且已证实在南美患者中存在源自西班牙的奠基者突变。
我们描述了一个哥伦比亚家族中Q163X突变的物理和组织学特征以及分子影响。
我们报告了两名女性患者,她们是近亲结婚父母的女儿,在出生后的头两年内出现症状,发展为严重的功能障碍,没有畸形特征、声音嘶哑或膈肌麻痹的迹象。电生理测试显示为轴索性感觉和运动神经病变。我们对GDAP1基因进行了测序,研究在第4外显子中鉴定出一个纯合点突变(c.487 C>T),导致过早出现终止密码子(p.Q163X)。这一结果证实了4A型腓骨肌萎缩症的诊断。
患者被转介至物理医学与康复科进行行走辅助评估。呼吸科对她们进行了随访,以评估肺功能和膈肌麻痹情况。我们提供了遗传咨询。患者的家谱研究、表型特征和电生理结果必须作为腓骨肌萎缩症患者临床诊断中的重要工具,以便确定致病突变。对于南美裔患者,应考虑GDAP1基因突变的存在,尤其是Q163X突变,作为CMT4A疾病的病因。