Li Peng, Zhang Lina, Zhang Mei, Zhou Changyong, Lin Nan
The Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministy of Education and Chinese Ministy of Public Health, Qilu Hospital of Shandong University, Jinan, Shandong, P.R. China.
Department of Ophthalmology, The Affiliated Hospital of Qingdao University, Qingdao, Shandong, P.R. China.
Int J Mol Med. 2016 Apr;37(4):989-97. doi: 10.3892/ijmm.2016.2491. Epub 2016 Feb 18.
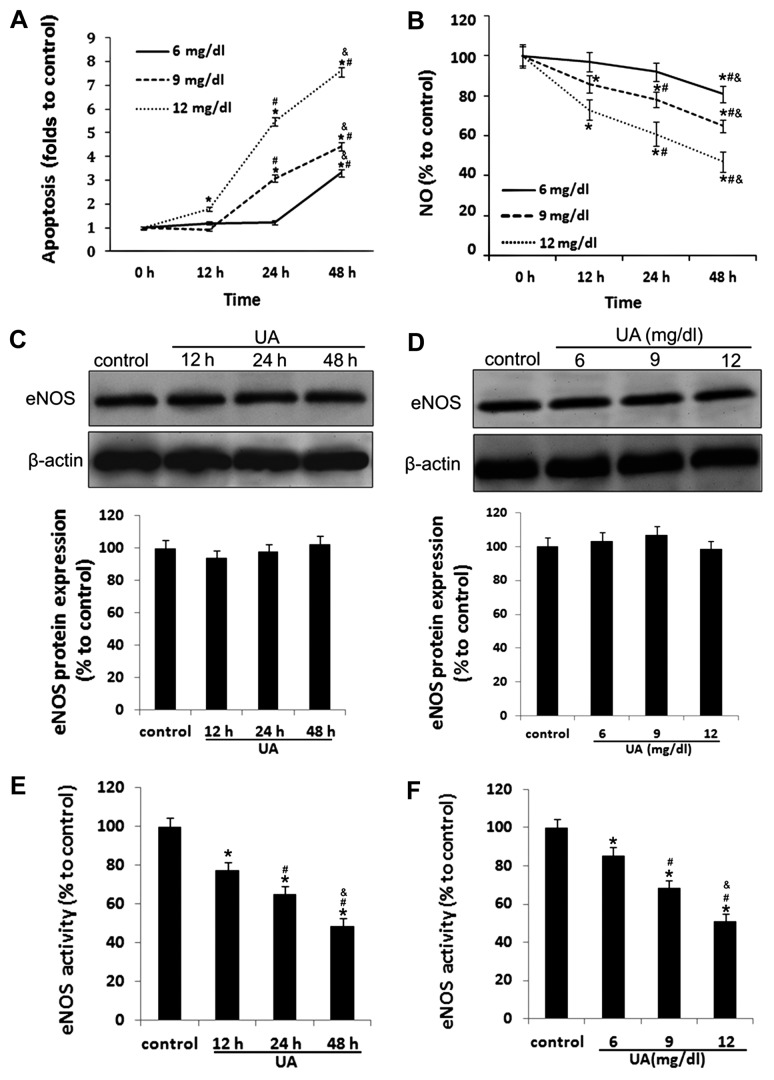
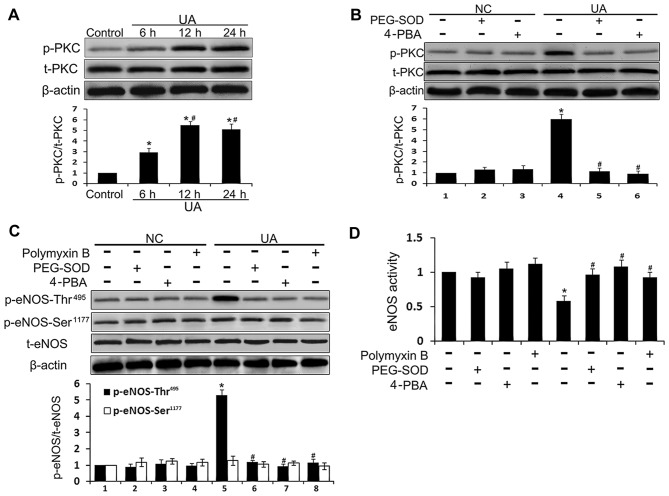
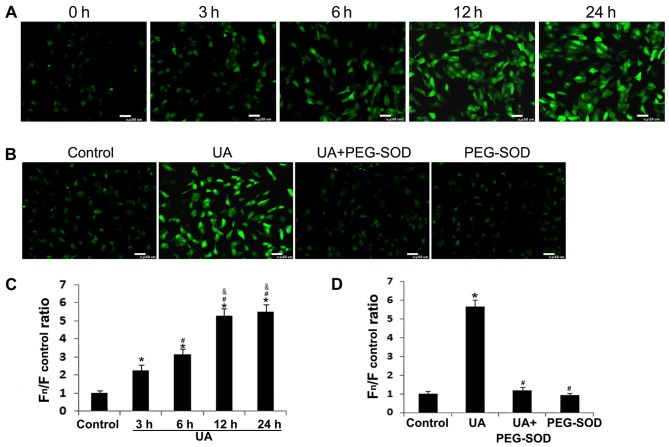
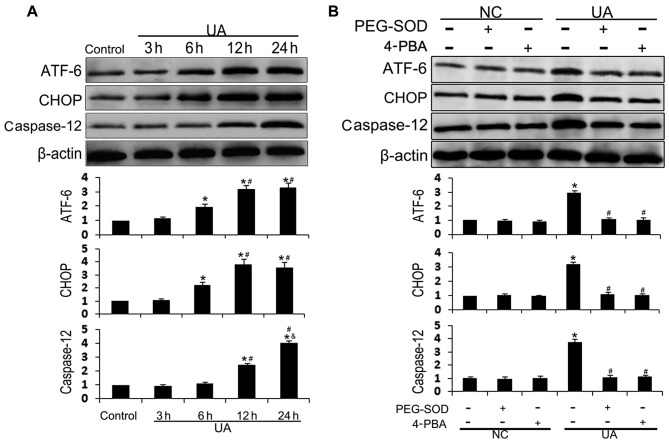
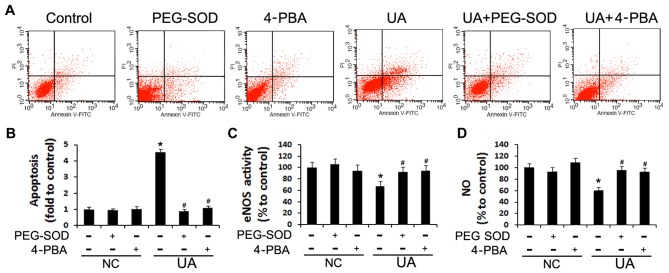
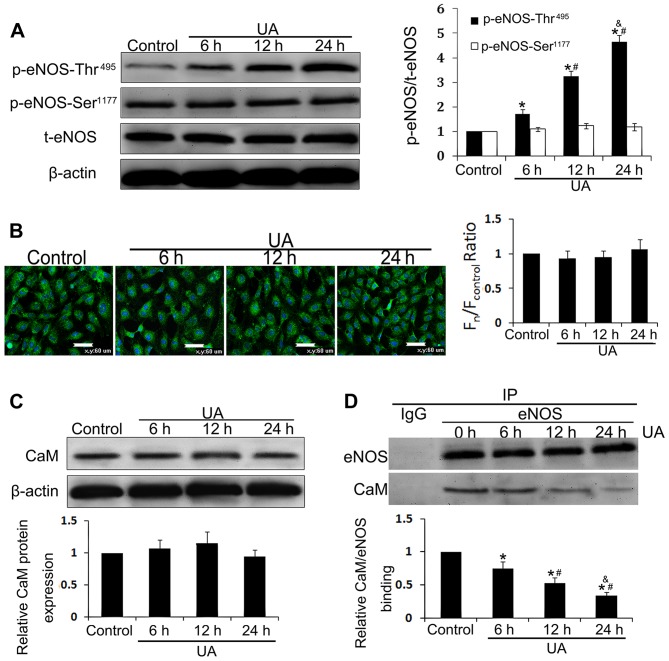
The mechanism by which hyperuricemia induced-endothelial dysfunction contributes to cardiovascular diseases (CVDs) is not yet fully understood. In the present study, we used uric acid (UA) to trigger endothelial dysfunction in cultured endothelial cells, and investigated the effects of induced reactive oxygen species (ROS) generation, endoplasmic reticulum (ER) stress induction, and the protein kinase C (PKC)-dependent endothelial nitric oxide synthase (eNOS) signaling pathway. Human umbilical vein endothelial cells (HUVECs) were incubated with 6, 9 or 12 mg/dl UA, ROS scavenger polyethylene glycol-superoxide dismutase (PEG‑SOD), ER stress inhibitor 4-phenylbutyric acid (4-PBA), and PKC inhibitor polymyxin B for 6-48 h. Nitric oxide (NO) production, eNOS activity, intracellular ROS, ER stress levels, and the interaction between eNOS and calmodulin (CaM) and cytosolic calcium levels were assessed using fluorescence microscopy and western blot analysis. Apoptosis was assessed by annexin V staining. UA increased HUVEC apoptosis and reduced eNOS activity and NO production in a dose- and time-dependent manner. Intracellular ROS was elevated after 3 h, while ER stress level increased after 6 h. UA did not alter intracellular Ca2+, CaM, or eNOS concentration, or eNOS Ser1177 phosphorylation. However, PKC-dependent eNOS phosphorylation at Thr495 was greatly enhanced, and consequently interaction between eNOS and CaM was reduced. Cellular ROS depletion, ER stress inhibition and PKC activity reduction inhibited the effect of UA on eNOS activity, NO release and apoptosis in HUVECs. Thus, we concluded that UA induced HUVEC apoptosis and endothelial dysfunction by triggering oxidative and ER stress through PKC/eNOS-mediated eNOS activity and NO production.
高尿酸血症诱导的内皮功能障碍导致心血管疾病(CVDs)的机制尚未完全阐明。在本研究中,我们使用尿酸(UA)诱导培养的内皮细胞发生功能障碍,并研究诱导活性氧(ROS)生成、内质网(ER)应激诱导以及蛋白激酶C(PKC)依赖性内皮型一氧化氮合酶(eNOS)信号通路的作用。将人脐静脉内皮细胞(HUVECs)与6、9或12mg/dl的UA、ROS清除剂聚乙二醇超氧化物歧化酶(PEG-SOD)、ER应激抑制剂4-苯基丁酸(4-PBA)和PKC抑制剂多粘菌素B孵育6-48小时。使用荧光显微镜和蛋白质印迹分析评估一氧化氮(NO)生成、eNOS活性、细胞内ROS、ER应激水平以及eNOS与钙调蛋白(CaM)之间的相互作用和胞质钙水平。通过膜联蛋白V染色评估细胞凋亡。UA以剂量和时间依赖性方式增加HUVEC凋亡并降低eNOS活性和NO生成。3小时后细胞内ROS升高,6小时后ER应激水平增加。UA未改变细胞内Ca2+、CaM或eNOS浓度,也未改变eNOS Ser1177磷酸化。然而,PKC依赖性eNOS在Thr495处的磷酸化大大增强,因此eNOS与CaM之间的相互作用减少。细胞内ROS消耗、ER应激抑制和PKC活性降低抑制了UA对HUVECs中eNOS活性、NO释放和细胞凋亡的影响。因此,我们得出结论,UA通过PKC/eNOS介导的eNOS活性和NO生成引发氧化应激和ER应激,从而诱导HUVEC凋亡和内皮功能障碍。