Forster Dominik, Dunthorn Micah, Stoeck Thorsten, Mahé Frédéric
Department of Ecology, Technische Universität Kaiserslautern , Kaiserslautern , Germany.
PeerJ. 2016 Feb 25;4:e1692. doi: 10.7717/peerj.1692. eCollection 2016.
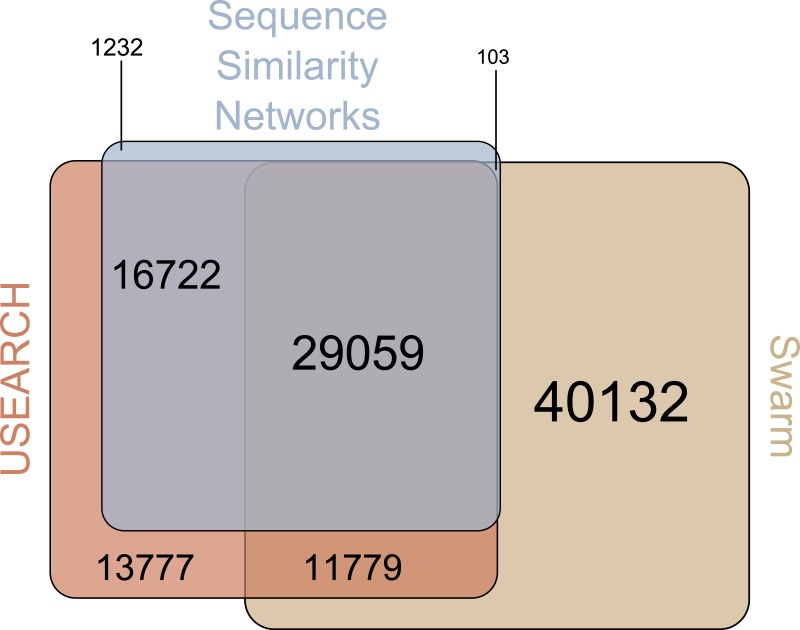
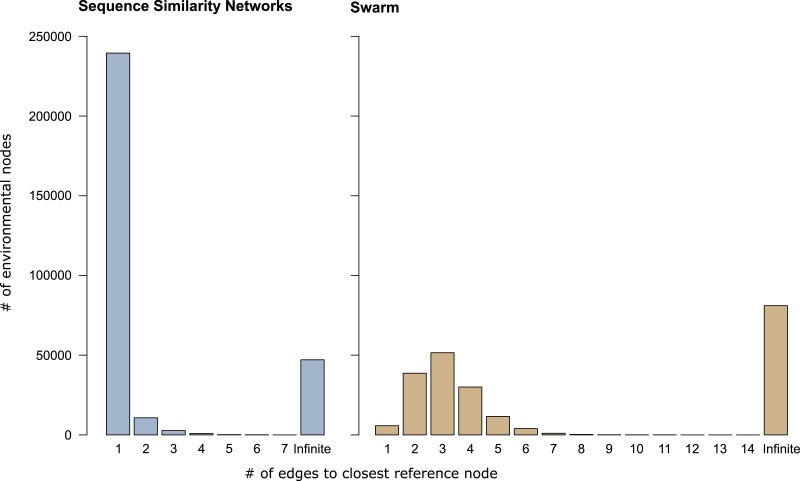
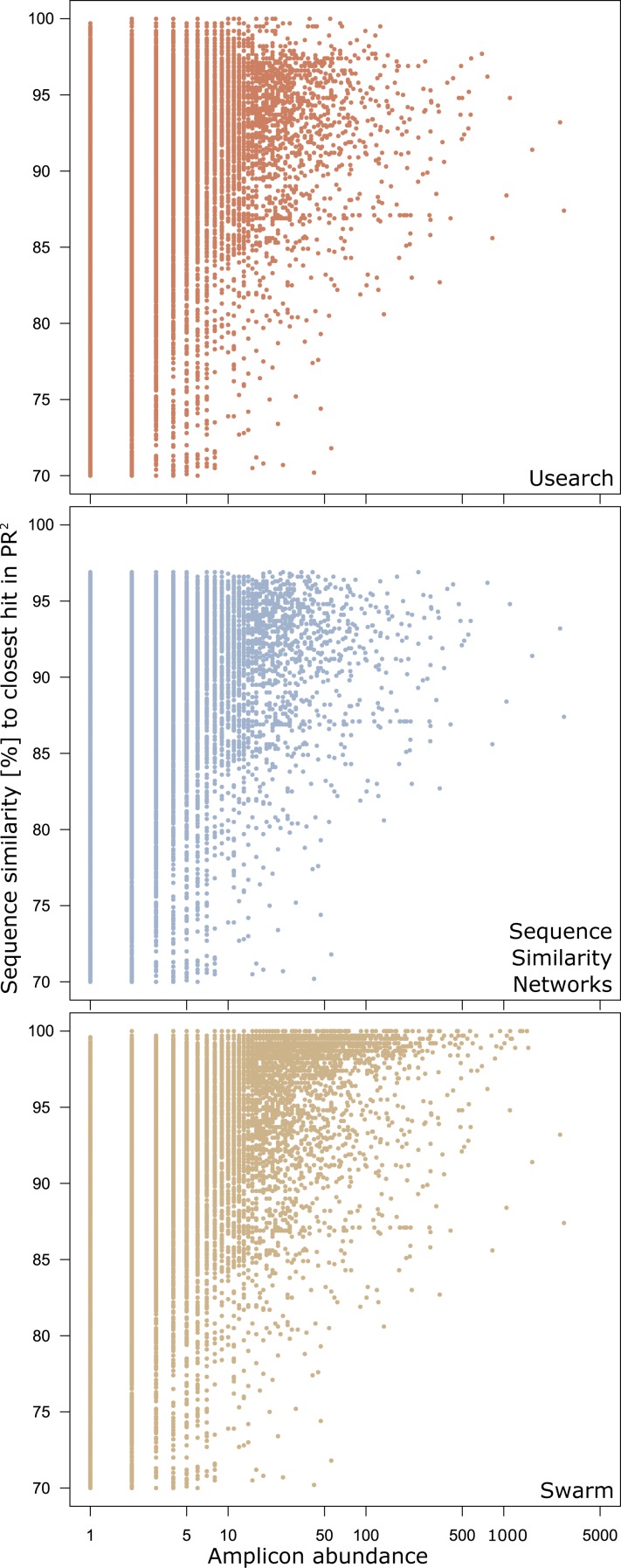
Discovery of novel diversity in high-throughput sequencing studies is an important aspect in environmental microbial ecology. To evaluate the effects that amplicon clustering methods have on the discovery of novel diversity, we clustered an environmental marine high-throughput sequencing dataset of protist amplicons together with reference sequences from the taxonomically curated Protist Ribosomal Reference (PR(2)) database using three de novo approaches: sequence similarity networks, USEARCH, and Swarm. The potentially novel diversity uncovered by each clustering approach differed drastically in the number of operational taxonomic units (OTUs) and in the number of environmental amplicons in these novel diversity OTUs. Global pairwise alignment comparisons revealed that numerous amplicons classified as potentially novel by USEARCH and Swarm were more than 97% similar to references of PR(2). Using shortest path analyses on sequence similarity network OTUs and Swarm OTUs we found additional novel diversity within OTUs that would have gone unnoticed without further exploiting their underlying network topologies. These results demonstrate that graph theory provides powerful tools for microbial ecology and the analysis of environmental high-throughput sequencing datasets. Furthermore, sequence similarity networks were most accurate in delineating novel diversity from previously discovered diversity.
在高通量测序研究中发现新的多样性是环境微生物生态学的一个重要方面。为了评估扩增子聚类方法对新多样性发现的影响,我们使用三种从头开始的方法,即序列相似性网络、USEARCH和Swarm,将一个原生生物扩增子的环境海洋高通量测序数据集与来自分类学整理的原生生物核糖体参考(PR(2))数据库的参考序列进行聚类。每种聚类方法发现的潜在新多样性在操作分类单元(OTU)数量以及这些新多样性OTU中的环境扩增子数量方面差异巨大。全局成对比对比较显示,许多被USEARCH和Swarm分类为潜在新序列的扩增子与PR(2)的参考序列相似度超过97%。通过对序列相似性网络OTU和Swarm OTU进行最短路径分析,我们在OTU内发现了额外的新多样性,如果不进一步利用其潜在的网络拓扑结构,这些多样性可能会被忽视。这些结果表明,图论为微生物生态学和环境高通量测序数据集的分析提供了强大的工具。此外,在区分新发现的多样性和先前发现的多样性方面,序列相似性网络最为准确。