Latger-Cannard Veronique, Philippe Christophe, Bouquet Alexandre, Baccini Veronique, Alessi Marie-Christine, Ankri Annick, Bauters Anne, Bayart Sophie, Cornillet-Lefebvre Pascale, Daliphard Sylvie, Mozziconacci Marie-Joelle, Renneville Aline, Ballerini Paola, Leverger Guy, Sobol Hagay, Jonveaux Philippe, Preudhomme Claude, Nurden Paquita, Lecompte Thomas, Favier Remi
Service d'Hématologie Biologique, Centre Hospitalier Universitaire de Nancy, Nancy, France.
Centre de Compétence Nord-Est des Pathologies Plaquettaires (CCPP), Nancy, France.
Orphanet J Rare Dis. 2016 Apr 26;11:49. doi: 10.1186/s13023-016-0432-0.
Less than 50 patients with FPD/AML (OMIM 601309) have been reported as of today and there may an underestimation. The purpose of this study was to describe the natural history, the haematological features and the genotype-phenotype correlations of this entity in order to, first, screen it better and earlier, before leukaemia occurrence and secondly to optimize appropriate monitoring and treatment, in particular when familial stem cell transplantation is considered.
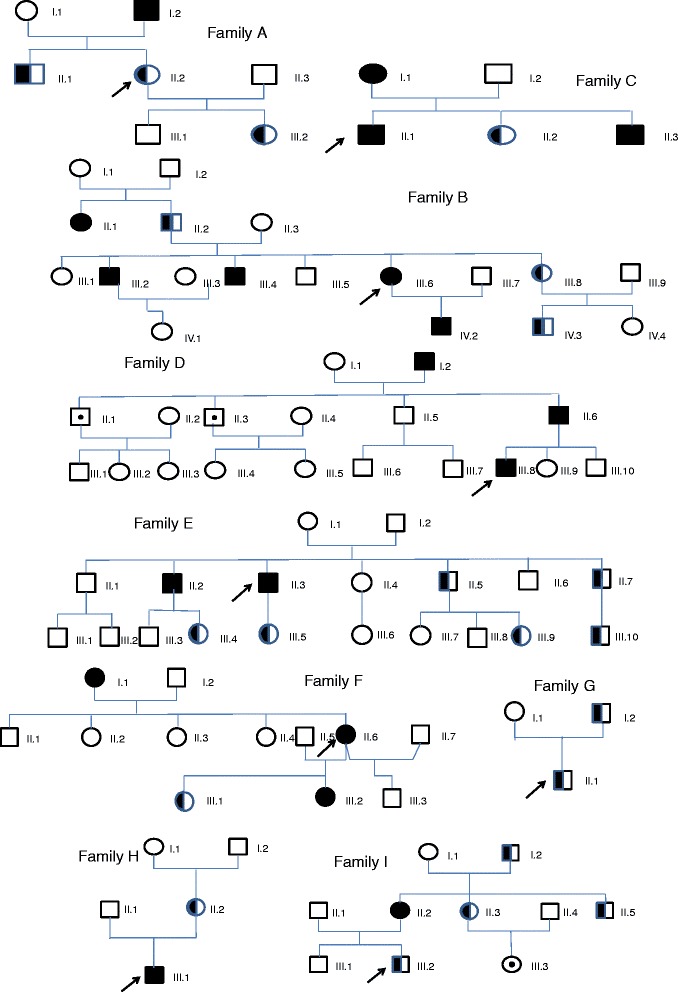
We have investigated 41 carriers of RUNX1 alteration belonging to nine unrelated French families with FPD/AML and two syndromic patients, registered in the French network on rare platelet disorders from 2005 to 2015.
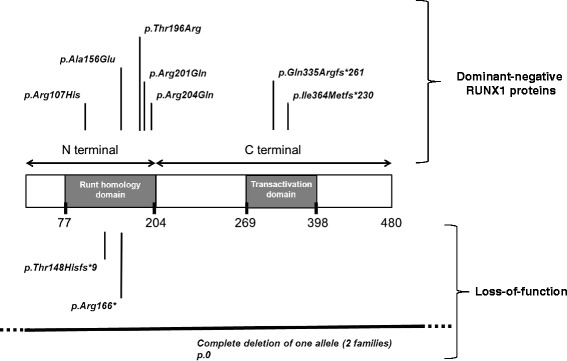
Five missense, one non-sense, three frameshift mutations and two large deletions involving several genes including RUNX1 were evidenced. The history of familial leukaemia was suggestive of FPD/AML in seven pedigrees, whereas an autosomal dominant pattern of lifelong thrombocytopenia was the clinical presentation of two. Additional syndromic features characterized two large sporadic deletions. Bleeding tendency was mild and thrombocytopenia moderate (>50 x10(9)/L), with normal platelet volume. A functional platelet defect consistent with a δ-granule release defect was found in ten patients regardless of the type of RUNX1 alteration. The incidence of haematological malignancies was higher when the mutated RUNX1 allele was likely to cause a dominant negative effect (19/34) in comparison with loss of function alleles (3/9). A normal platelet count does not rule out the diagnosis of FPD/AML, since the platelet count was found normal for three mutated subjects, a feature that has a direct impact in the search for a related donor in case of allogeneic haematopoietic stem cell transplantation.
Platelet dysfunction suggestive of defective δ-granule release could be of values for the diagnosis of FPD/AML particularly when the clinical presentation is an autosomal dominant thrombocytopenia with normal platelet size in the absence of familial malignancies. The genotype-phenotype correlations might be helpful in genetic counselling and appropriate optimal therapeutic management.
截至目前,报道的家族性血小板减少伴骨髓增生异常综合征/急性髓系白血病(FPD/AML,OMIM 601309)患者不足50例,实际病例数可能被低估。本研究旨在描述该疾病的自然史、血液学特征以及基因型-表型相关性,以便首先在白血病发生前更好、更早地进行筛查,其次优化适当的监测和治疗,特别是在考虑进行家族性干细胞移植时。
我们调查了2005年至2015年在法国罕见血小板疾病网络登记的9个不相关的法国FPD/AML家族中的41名RUNX1基因改变携带者以及2名综合征患者。
发现了5个错义突变、1个无义突变、3个移码突变以及2个涉及包括RUNX1在内多个基因的大片段缺失。7个家系的家族性白血病病史提示为FPD/AML,而2个家系的临床表现为常染色体显性遗传的终身血小板减少症。另外,2个大片段散发性缺失具有额外的综合征特征。出血倾向较轻,血小板减少程度中等(>50×10⁹/L),血小板体积正常。无论RUNX1改变的类型如何,10例患者均发现存在与δ颗粒释放缺陷一致的功能性血小板缺陷。与功能缺失等位基因(3/9)相比,当突变的RUNX1等位基因可能导致显性负效应时(19/34),血液系统恶性肿瘤的发生率更高。血小板计数正常并不能排除FPD/AML的诊断,因为3名突变受试者的血小板计数正常,这一特征在同种异体造血干细胞移植时寻找相关供体方面有直接影响。
提示δ颗粒释放缺陷的血小板功能障碍对FPD/AML的诊断可能有价值,特别是当临床表现为常染色体显性遗传的血小板减少症且血小板大小正常且无家族性恶性肿瘤时。基因型-表型相关性可能有助于遗传咨询和适当的最佳治疗管理。