Knoppova Barbora, Reily Colin, Maillard Nicolas, Rizk Dana V, Moldoveanu Zina, Mestecky Jiri, Raska Milan, Renfrow Matthew B, Julian Bruce A, Novak Jan
Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, USA; Department of Immunology, Faculty of Medicine and Dentistry, Palacky University and University Hospital, Olomouc, Czech Republic.
Department of Medicine, University of Alabama at Birmingham , Birmingham, AL , USA.
Front Immunol. 2016 Apr 12;7:117. doi: 10.3389/fimmu.2016.00117. eCollection 2016.
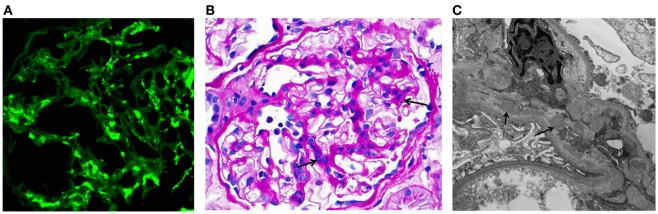
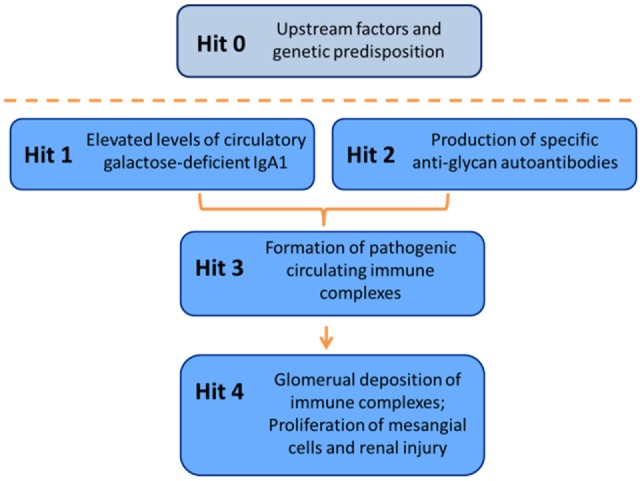
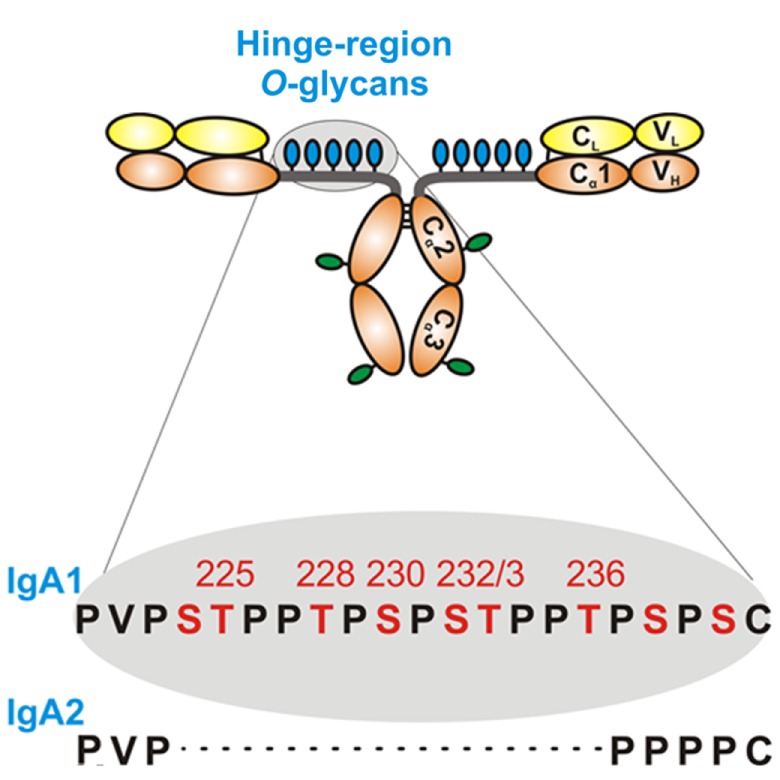
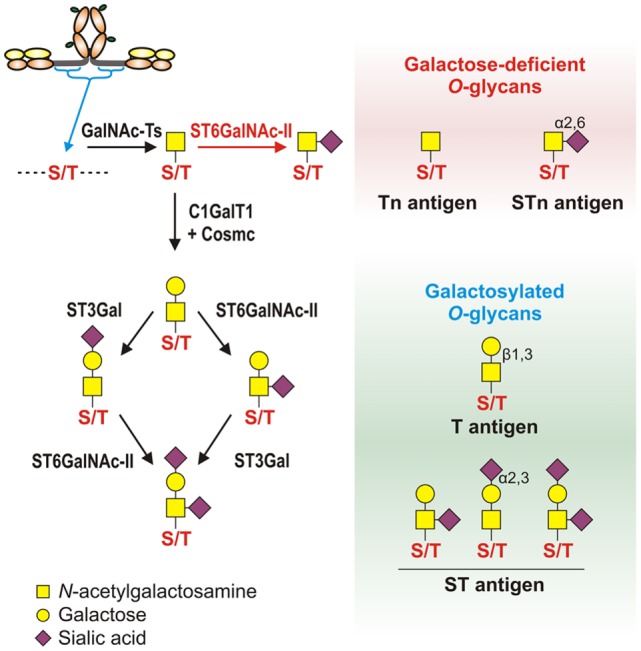
IgA nephropathy (IgAN) is the most common primary glomerulonephritis, frequently leading to end-stage renal disease, as there is no disease-specific therapy. IgAN is diagnosed from pathological assessment of a renal biopsy specimen based on predominant or codominant IgA-containing immunodeposits, usually with complement C3 co-deposits and with variable presence of IgG and/or IgM. The IgA in these renal deposits is galactose-deficient IgA1, with less than a full complement of galactose residues on the O-glycans in the hinge region of the heavy chains. Research from the past decade led to the definition of IgAN as an autoimmune disease with a multi-hit pathogenetic process with contributing genetic and environmental components. In this process, circulating galactose-deficient IgA1 (autoantigen) is bound by antiglycan IgG or IgA (autoantibodies) to form immune complexes. Some of these circulating complexes deposit in glomeruli, and thereby activate mesangial cells and induce renal injury through cellular proliferation and overproduction of extracellular matrix components and cytokines/chemokines. Glycosylation pathways associated with production of the autoantigen and the unique characteristics of the corresponding autoantibodies in patients with IgAN have been uncovered. Complement likely plays a significant role in the formation and the nephritogenic activities of these complexes. Complement activation is mediated through the alternative and lectin pathways and probably occurs systemically on IgA1-containing circulating immune complexes as well as locally in glomeruli. Incidence of IgAN varies greatly by geographical location; the disease is rare in central Africa but accounts for up to 40% of native-kidney biopsies in eastern Asia. Some of this variation may be explained by genetically determined influences on the pathogenesis of the disease. Genome-wide association studies to date have identified several loci associated with IgAN. Some of these loci are associated with the increased prevalence of IgAN, whereas others, such as deletion of complement factor H-related genes 1 and 3, are protective against the disease. Understanding the molecular mechanisms and genetic and biochemical factors involved in formation and activities of pathogenic IgA1-containing immune complexes will enable the development of future disease-specific therapies as well as identification of non-invasive disease-specific biomarkers.
IgA 肾病(IgAN)是最常见的原发性肾小球肾炎,由于缺乏针对该疾病的特异性治疗方法,常常会发展为终末期肾病。IgAN 通过对肾活检标本进行病理评估来诊断,依据主要或共显性含 IgA 的免疫沉积物,通常伴有补体 C3 共沉积,且 IgG 和/或 IgM 存在情况各异。这些肾沉积物中的 IgA 是半乳糖缺陷型 IgA1,其重链铰链区 O - 聚糖上的半乳糖残基不完全。过去十年的研究将 IgAN 定义为一种自身免疫性疾病,具有多因素致病过程,涉及遗传和环境因素。在此过程中,循环中的半乳糖缺陷型 IgA1(自身抗原)与抗聚糖 IgG 或 IgA(自身抗体)结合形成免疫复合物。其中一些循环复合物沉积在肾小球中,从而激活系膜细胞,并通过细胞增殖以及细胞外基质成分和细胞因子/趋化因子的过度产生诱导肾损伤。与 IgAN 患者自身抗原产生相关的糖基化途径以及相应自身抗体的独特特征已被揭示。补体可能在这些复合物的形成和致肾炎活性中起重要作用。补体激活通过替代途径和凝集素途径介导,可能在含 IgA1 的循环免疫复合物上全身性发生,也在肾小球局部发生。IgAN 的发病率因地理位置差异很大;该疾病在中非罕见,但在东亚占肾活检的比例高达 40%。这种差异部分可能由遗传决定的对疾病发病机制的影响来解释。迄今为止的全基因组关联研究已确定了几个与 IgAN 相关的基因座。其中一些基因座与 IgAN 患病率增加相关,而其他一些,如补体因子 H 相关基因 1 和 3 的缺失,则对该疾病有保护作用。了解参与致病性含 IgA1 免疫复合物形成和活性的分子机制以及遗传和生化因素,将有助于开发未来针对该疾病的特异性疗法,并识别非侵入性疾病特异性生物标志物。