López-Pérez Mario, Rodriguez-Valera Francisco
Evolutionary Genomics Group, Departamento de Producción Vegetal y Microbiología, Universidad Miguel Hernández, Alicante, Spain.
Evolutionary Genomics Group, Departamento de Producción Vegetal y Microbiología, Universidad Miguel Hernández, Alicante, Spain
Genome Biol Evol. 2016 Jun 3;8(5):1556-70. doi: 10.1093/gbe/evw098.
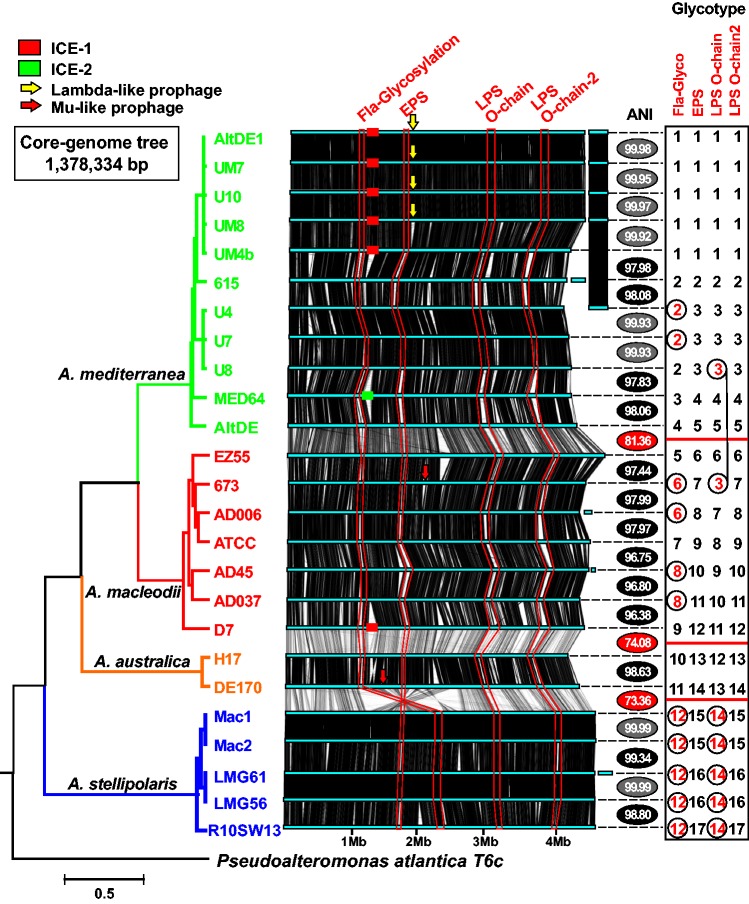
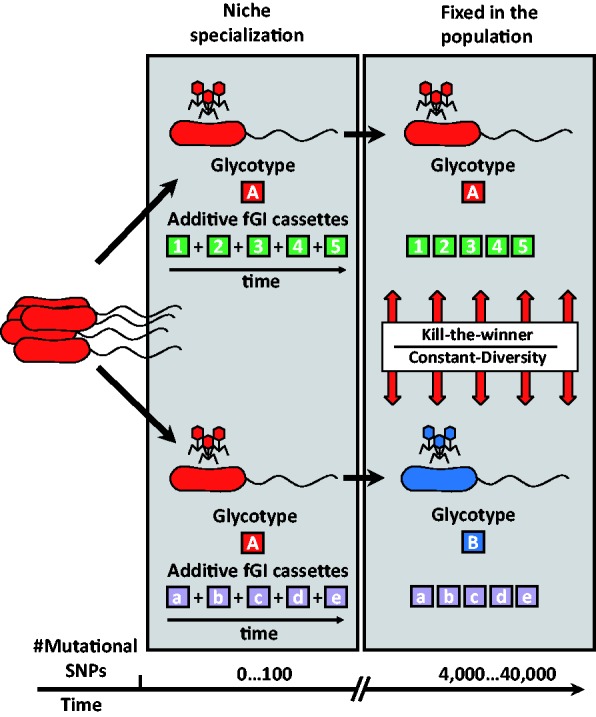
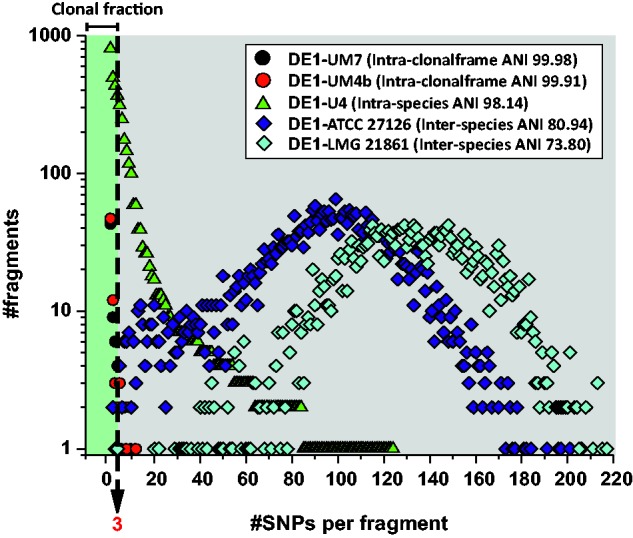
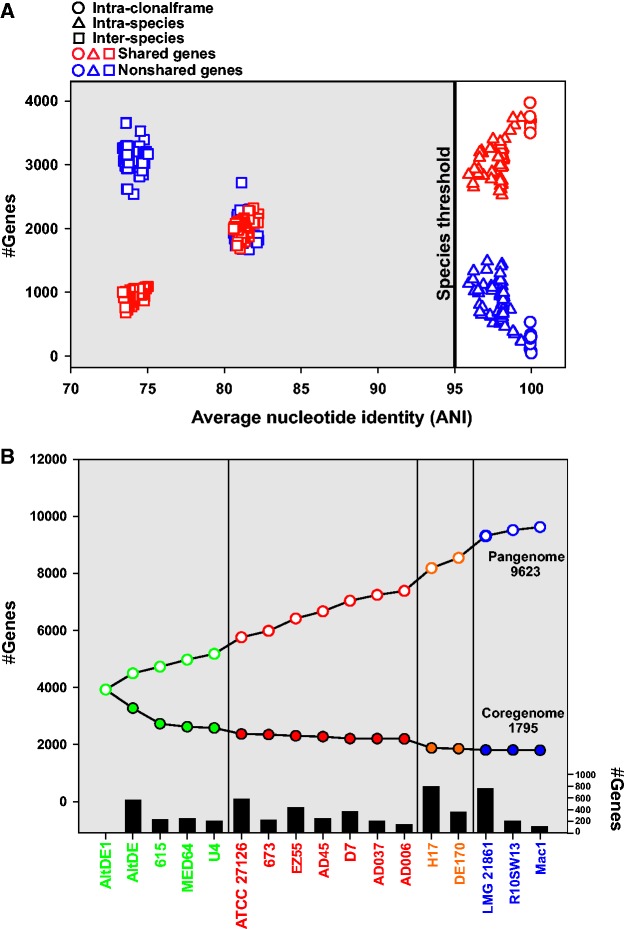
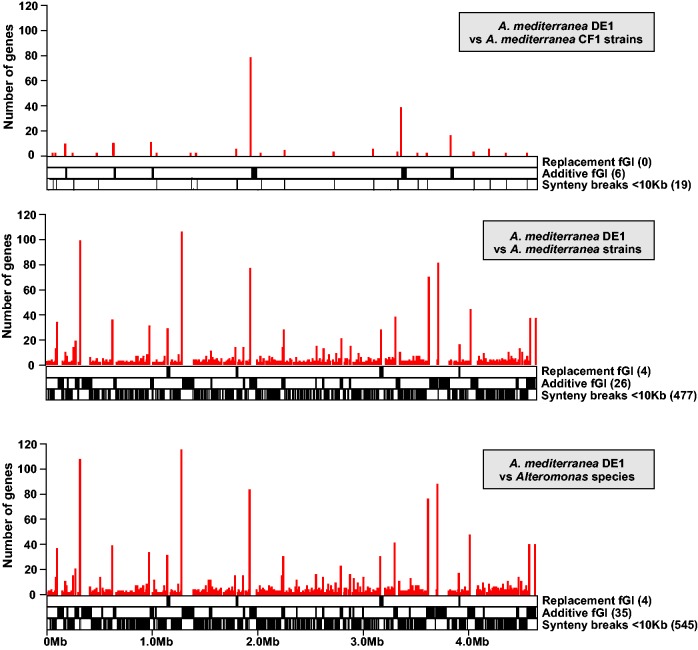
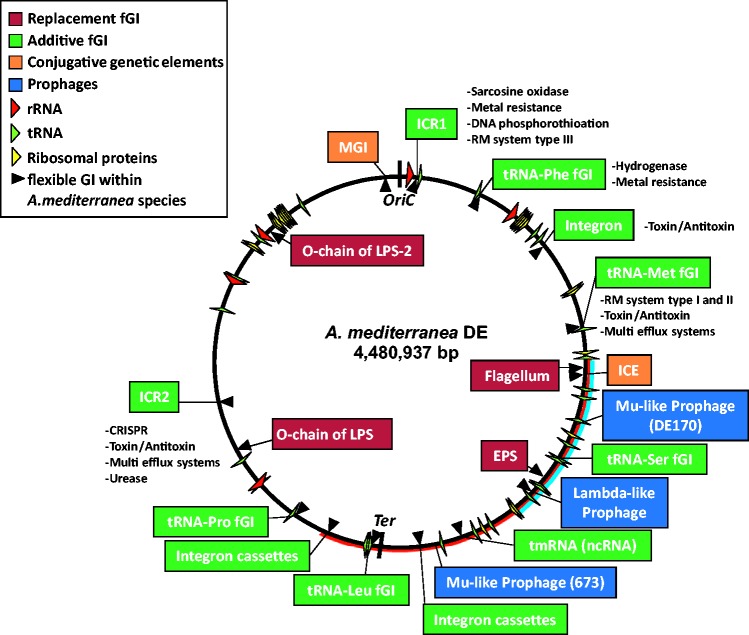
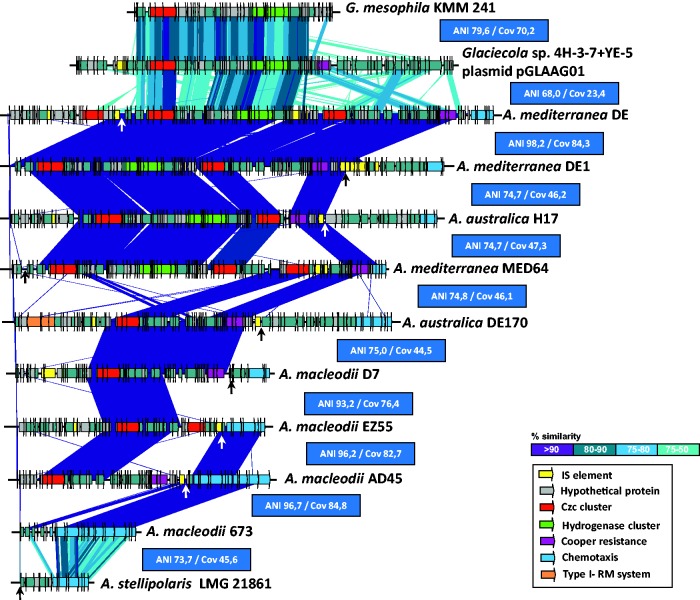
We have examined a collection of the free-living marine bacterium Alteromonas genomes with cores diverging in average nucleotide identities ranging from 99.98% to 73.35%, i.e., from microbes that can be considered members of a natural clone (like in a clinical epidemiological outbreak) to borderline genus level. The genomes were largely syntenic allowing a precise delimitation of the core and flexible regions in each. The core was 1.4 Mb (ca. 30% of the typical strain genome size). Recombination rates along the core were high among strains belonging to the same species (37.7-83.7% of all nucleotide polymorphisms) but they decreased sharply between species (18.9-5.1%). Regarding the flexible genome, its main expansion occurred within the boundaries of the species, i.e., strains of the same species already have a large and diverse flexible genome. Flexible regions occupy mostly fixed genomic locations. Four large genomic islands are involved in the synthesis of strain-specific glycosydic receptors that we have called glycotypes. These genomic regions are exchanged by homologous recombination within and between species and there is evidence for their import from distant taxonomic units (other genera within the family). In addition, several hotspots for integration of gene cassettes by illegitimate recombination are distributed throughout the genome. They code for features that give each clone specific properties to interact with their ecological niche and must flow fast throughout the whole genus as they are found, with nearly identical sequences, in different species. Models for the generation of this genomic diversity involving phage predation are discussed.
我们研究了一组自由生活的海洋细菌交替单胞菌属的基因组,其核心区域的平均核苷酸同一性差异范围为99.98%至73.35%,即从可被视为自然克隆成员的微生物(如临床流行病学爆发中的情况)到接近属水平。这些基因组在很大程度上是同线的,从而能够精确界定每个基因组中的核心区域和可变区域。核心区域为1.4 Mb(约占典型菌株基因组大小的30%)。在属于同一物种的菌株中,核心区域的重组率很高(占所有核苷酸多态性的37.7 - 83.7%),但在不同物种之间则急剧下降(18.9 - 5.1%)。关于可变基因组,其主要扩展发生在物种界限内,即同一物种的菌株已经拥有庞大且多样的可变基因组。可变区域大多占据固定的基因组位置。四个大的基因组岛参与了菌株特异性糖苷受体的合成,我们将其称为糖型。这些基因组区域通过种内和种间的同源重组进行交换,并且有证据表明它们是从遥远的分类单元(该科内的其他属)导入的。此外,通过非法重组整合基因盒的几个热点分布在整个基因组中。它们编码的特征赋予每个克隆与生态位相互作用的特定属性,并且由于在不同物种中发现具有几乎相同的序列,这些特征必须在整个属中快速传播。文中讨论了涉及噬菌体捕食的这种基因组多样性产生的模型。