Oetjens Matthew T, Brown-Gentry Kristin, Goodloe Robert, Dilks Holli H, Crawford Dana C
Center for Human Genetics Research Vanderbilt University, Nashville TN, USA.
Sarah Cannon Research Institute, Nashville TN, USA.
Front Genet. 2016 May 6;7:76. doi: 10.3389/fgene.2016.00076. eCollection 2016.
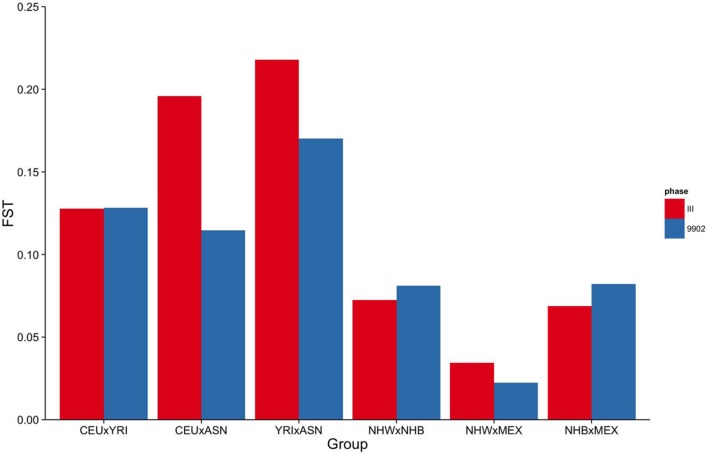
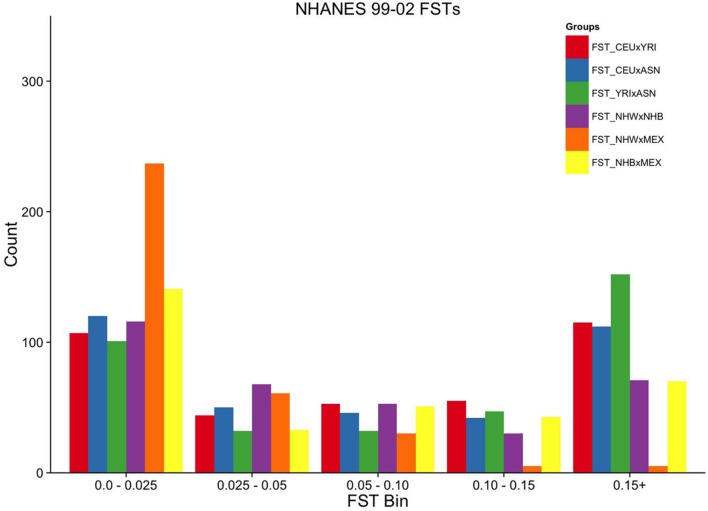
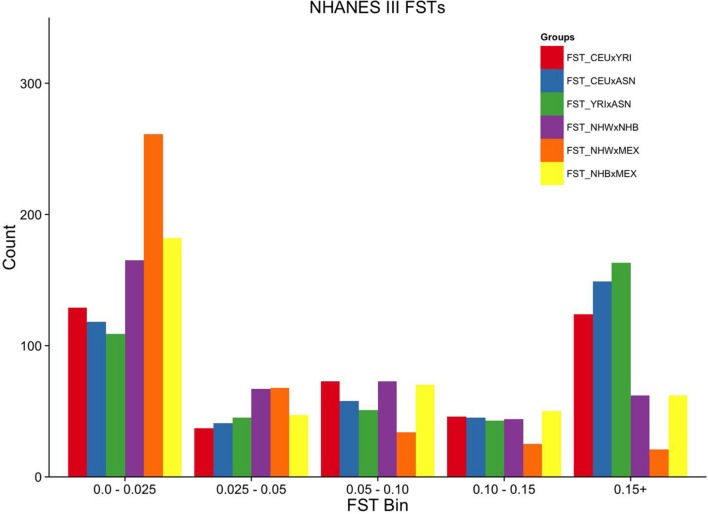
Population stratification or confounding by genetic ancestry is a potential cause of false associations in genetic association studies. Estimation of and adjustment for genetic ancestry has become common practice thanks in part to the availability of ancestry informative markers on genome-wide association study (GWAS) arrays. While array data is now widespread, these data are not ubiquitous as several large epidemiologic and clinic-based studies lack genome-wide data. One such large epidemiologic-based study lacking genome-wide data accessible to investigators is the National Health and Nutrition Examination Surveys (NHANES), population-based cross-sectional surveys of Americans linked to demographic, health, and lifestyle data conducted by the Centers for Disease Control and Prevention. DNA samples (n = 14,998) were extracted from biospecimens from consented NHANES participants between 1991-1994 (NHANES III, phase 2) and 1999-2002 and represent three major self-identified racial/ethnic groups: non-Hispanic whites (n = 6,634), non-Hispanic blacks (n = 3,458), and Mexican Americans (n = 3,950). We as the Epidemiologic Architecture for Genes Linked to Environment study genotyped candidate gene and GWAS-identified index variants in NHANES as part of the larger Population Architecture using Genomics and Epidemiology I study for collaborative genetic association studies. To enable basic quality control such as estimation of genetic ancestry to control for population stratification in NHANES san genome-wide data, we outline here strategies that use limited genetic data to identify the markers optimal for characterizing genetic ancestry. From among 411 and 295 autosomal SNPs available in NHANES III and NHANES 1999-2002, we demonstrate that markers with ancestry information can be identified to estimate global ancestry. Despite limited resolution, global genetic ancestry is highly correlated with self-identified race for the majority of participants, although less so for ethnicity. Overall, the strategies outlined here for a large epidemiologic study can be applied to other datasets accessible for genotype-phenotype studies but are sans genome-wide data.
群体分层或基因血统混杂是基因关联研究中产生错误关联的一个潜在原因。对基因血统进行估计和调整已成为常规做法,这在一定程度上要归功于全基因组关联研究(GWAS)阵列上可获取的血统信息标记。虽然阵列数据现在很普遍,但这些数据并非无处不在,因为一些大型的基于流行病学和临床的研究缺乏全基因组数据。调查人员无法获取全基因组数据的一项此类大型基于流行病学的研究是美国国家健康与营养检查调查(NHANES),这是由疾病控制和预防中心开展的与人口统计学、健康和生活方式数据相关联的基于人群的美国人横断面调查。从1991年至1994年(NHANES III,第2阶段)以及1999年至2002年同意参与NHANES的参与者的生物样本中提取了DNA样本(n = 14,998),这些样本代表了三个主要的自我认定的种族/族裔群体:非西班牙裔白人(n = 6,634)、非西班牙裔黑人(n = 3,458)和墨西哥裔美国人(n = 3,950)。作为“与环境相关基因的流行病学架构”研究,我们对NHANES中的候选基因和GWAS识别的索引变体进行基因分型,这是更大规模的“利用基因组学和流行病学进行群体架构I”研究的一部分,用于合作性基因关联研究。为了在没有全基因组数据的NHANES中进行基本的质量控制,如估计基因血统以控制群体分层,我们在此概述了利用有限基因数据来识别最适合表征基因血统的标记的策略。从NHANES III和1999 - 2002年NHANES中可用的411个和295个常染色体单核苷酸多态性中,我们证明可以识别出具有血统信息的标记来估计全球血统。尽管分辨率有限,但对于大多数参与者来说,全球基因血统与自我认定的种族高度相关,尽管与族裔的相关性较低。总体而言,这里概述的针对大型流行病学研究的策略可应用于其他可用于基因型 - 表型研究但没有全基因组数据的数据集。