Palumbo Pietro, Palumbo Orazio, Leone Maria Pia, Stallone Raffaella, Palladino Teresa, Zelante Leopoldo, Carella Massimo
Laboratorio di Genetica Medica, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG), Italy.
Laboratorio di Genetica Medica, IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (FG), Italy ; Dipartimento di Scienze del suolo, della pianta e degli alimenti, Università degli Studi di Bari "Aldo Moro", Bari, Italy.
Mol Cytogenet. 2016 May 27;9:40. doi: 10.1186/s13039-016-0252-x. eCollection 2016.
Structural rearrangements of chromosome 19p13.3 are a rare condition, and their phenotypic consequences remain not well defined, because of the variability of clinical manifestations. Increasing knowledge of new 19p13.3 microdeletion is useful to clarify the phenotypic variability observed in some patients. In a small number of recent papers, patients with intellectual disabilities, multiple congenital anomalies and microdeletion of the chromosome band 19p13.3 have been described. However, little is known about genes responsible for clinical features in patients carriers of 19p13.3 microdeletion; thus, increasing number of reported cases will be helpful to investigate the contribution of candidate genes, providing bases for future investigations.
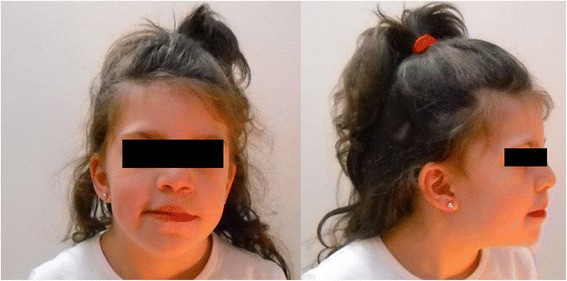
Here, we report on a 10-years-old girl referred to our genetics clinic due to intellectual disability, attention deficit, behavioral and speech delay, hypotonia, facial dysmorphisms, eye anomalies and congenital malformations. Using an high resolution SNP array, we identified a de novo microdeletion of chromosome 19p13.3, resulting in the heterozygous loss of 27 RefSeq genes and a miRNA, partially overlapping with three others deletions already reported in literature, but extending downstream (centromeric) for additional 386 Kb. This chromosomal region includes 13 genes amongst of which we suggest for the first time the APC2, PLK5 and MBD3 genes as potential functional candidates for neurodevelopmental and behavioral phenotypes observed.
Here we describe a patient with a 19p13.3 microdeletion that spans to the downstream chromosomal region with respect to the overlapping deletions previously reported in several other cases. The neurobehavioral features observed in our case has extended the phenotypic spectrum associated with the 19p13.3 microdeletion. New candidate genes are proposed for the neurobehavioral phenotype observed in our case.
19号染色体短臂1区3带(19p13.3)的结构重排是一种罕见病症,由于临床表现的变异性,其表型后果仍未得到很好的界定。对新发现的19p13.3微缺失的了解不断增加,有助于阐明在一些患者中观察到的表型变异性。在最近的少数几篇论文中,已经描述了患有智力残疾、多种先天性异常以及19p13.3染色体带微缺失的患者。然而,对于19p13.3微缺失患者临床特征的致病基因知之甚少;因此,增加报告病例数将有助于研究候选基因的作用,为未来的研究提供依据。
在此,我们报告一名10岁女孩,因智力残疾、注意力缺陷、行为和语言发育迟缓、肌张力减退、面部畸形、眼部异常和先天性畸形转诊至我们的遗传学诊所。使用高分辨率单核苷酸多态性(SNP)芯片,我们鉴定出19p13.3染色体的新生微缺失,导致27个RefSeq基因和一个微RNA(miRNA)杂合缺失,部分与文献中已报道的其他三个缺失重叠,但向中心粒方向下游延伸了另外386千碱基对(Kb)。这个染色体区域包含13个基因,其中我们首次提出腺瘤性息肉病蛋白2(APC2)、丝氨酸/苏氨酸蛋白激酶5(PLK5)和甲基-CpG结合结构域蛋白3(MBD3)基因作为观察到的神经发育和行为表型的潜在功能候选基因。
在此我们描述了一名患有19p13.3微缺失的患者,该缺失相对于先前在其他几例病例中报道的重叠缺失延伸至染色体下游区域。我们病例中观察到的神经行为特征扩展了与19p13.3微缺失相关的表型谱。针对我们病例中观察到的神经行为表型提出了新的候选基因。