van den Berg Irene, Boichard Didier, Guldbrandtsen Bernt, Lund Mogens S
Department of Molecular Biology and Genetics, Center for Quantitative Genetics and Genomics, Aarhus University, DK-8830 Tjele, Denmark Génétique Animale et Biologie Intégrative (GABI), French National Institute for Agricultural Research (INRA), AgroParisTech, Université Paris Saclay, 78350 Jouy-en-Josas, France
Génétique Animale et Biologie Intégrative (GABI), French National Institute for Agricultural Research (INRA), AgroParisTech, Université Paris Saclay, 78350 Jouy-en-Josas, France.
G3 (Bethesda). 2016 Aug 9;6(8):2553-61. doi: 10.1534/g3.116.027730.
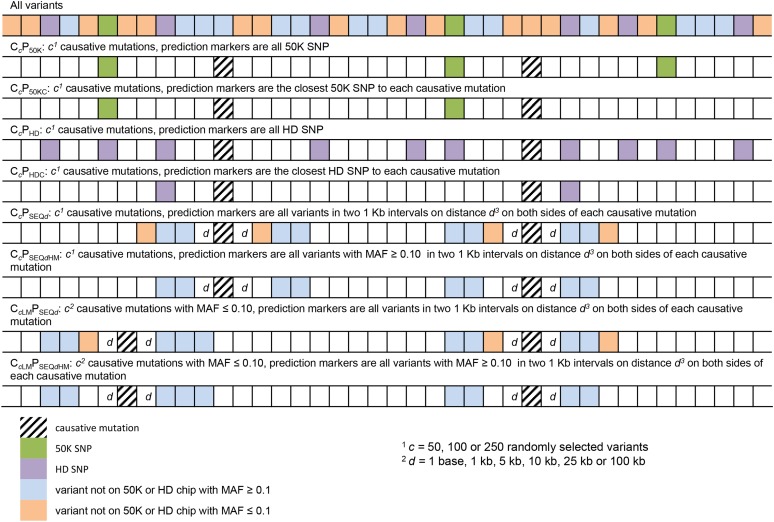
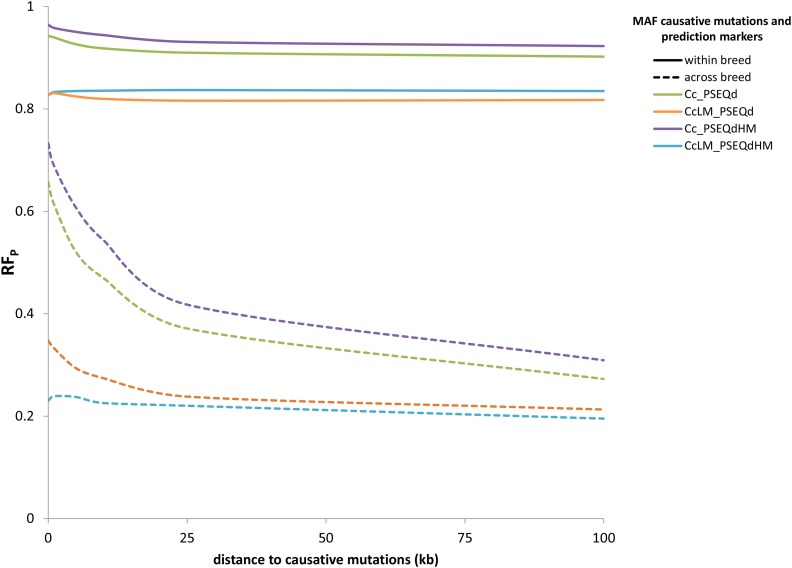
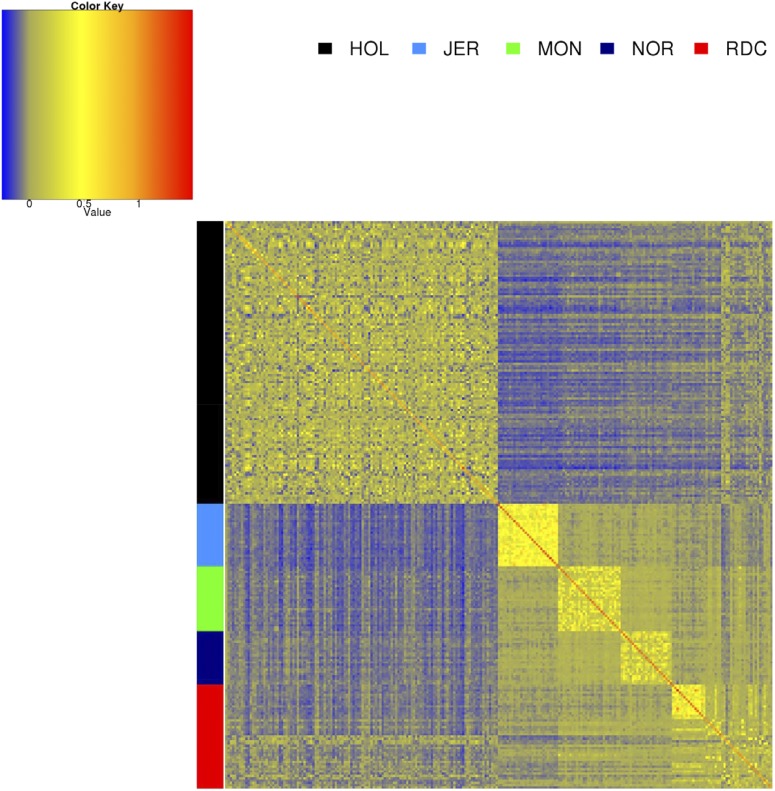
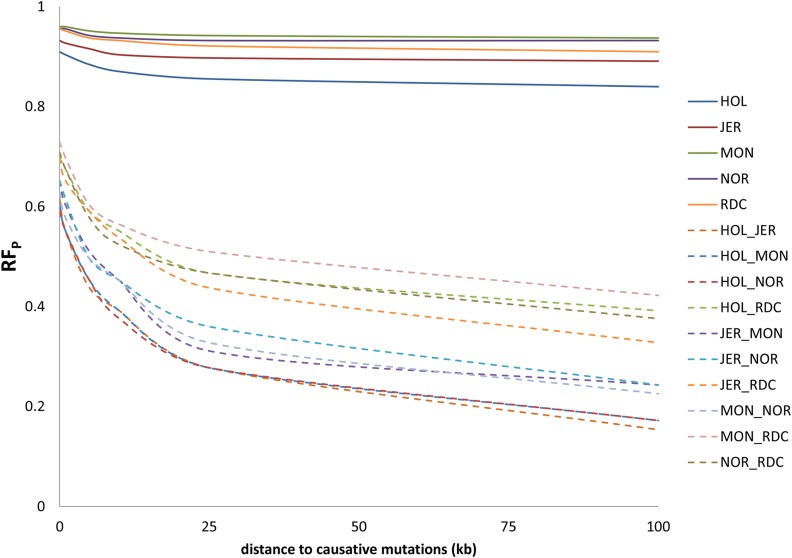
Sequence data are expected to increase the reliability of genomic prediction by containing causative mutations directly, especially in cases where low linkage disequilibrium between markers and causative mutations limits prediction reliability, such as across-breed prediction in dairy cattle. In practice, the causative mutations are unknown, and prediction with only variants in perfect linkage disequilibrium with the causative mutations is not realistic, leading to a reduced reliability compared to knowing the causative variants. Our objective was to use sequence data to investigate the potential benefits of sequence data for the prediction of genomic relationships, and consequently reliability of genomic breeding values. We used sequence data from five dairy cattle breeds, and a larger number of imputed sequences for two of the five breeds. We focused on the influence of linkage disequilibrium between markers and causative mutations, and assumed that a fraction of the causative mutations was shared across breeds and had the same effect across breeds. By comparing the loss in reliability of different scenarios, varying the distance between markers and causative mutations, using either all genome wide markers from commercial SNP chips, or only the markers closest to the causative mutations, we demonstrate the importance of using only variants very close to the causative mutations, especially for across-breed prediction. Rare variants improved prediction only if they were very close to rare causative mutations, and all causative mutations were rare. Our results show that sequence data can potentially improve genomic prediction, but careful selection of markers is essential.
序列数据有望通过直接包含致病突变来提高基因组预测的可靠性,特别是在标记与致病突变之间的连锁不平衡较低限制预测可靠性的情况下,例如奶牛的跨品种预测。在实际应用中,致病突变是未知的,仅使用与致病突变处于完全连锁不平衡状态的变异进行预测是不现实的,与知道致病变异相比,这会导致可靠性降低。我们的目标是利用序列数据研究序列数据在预测基因组关系以及基因组育种值可靠性方面的潜在益处。我们使用了五个奶牛品种的序列数据,以及五个品种中两个品种的大量推算序列。我们关注标记与致病突变之间的连锁不平衡的影响,并假设一部分致病突变在品种间共享且在品种间具有相同的效应。通过比较不同情况下可靠性的损失,改变标记与致病突变之间的距离,使用商业SNP芯片的全基因组标记或仅使用最接近致病突变的标记,我们证明了仅使用非常接近致病突变的变异的重要性,特别是对于跨品种预测。只有当稀有变异非常接近稀有致病突变且所有致病突变都是稀有时,稀有变异才能改善预测。我们的结果表明,序列数据有可能改善基因组预测,但仔细选择标记至关重要。