Thomson Emma, Ip Camilla L C, Badhan Anjna, Christiansen Mette T, Adamson Walt, Ansari M Azim, Bibby David, Breuer Judith, Brown Anthony, Bowden Rory, Bryant Josie, Bonsall David, Da Silva Filipe Ana, Hinds Chris, Hudson Emma, Klenerman Paul, Lythgow Kieren, Mbisa Jean L, McLauchlan John, Myers Richard, Piazza Paolo, Roy Sunando, Trebes Amy, Sreenu Vattipally B, Witteveldt Jeroen, Barnes Eleanor, Simmonds Peter
MRC-University of Glasgow Centre for Virus Research, Glasgow, United Kingdom.
Oxford Genomics Centre, Wellcome Trust Centre for Human Genetics, University of Oxford, Oxford, United Kingdom.
J Clin Microbiol. 2016 Oct;54(10):2470-84. doi: 10.1128/JCM.00330-16. Epub 2016 Jul 6.
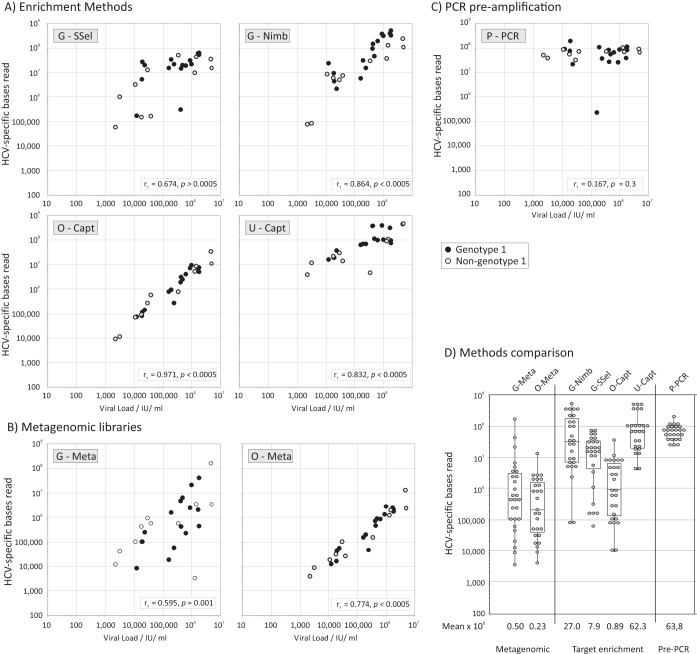
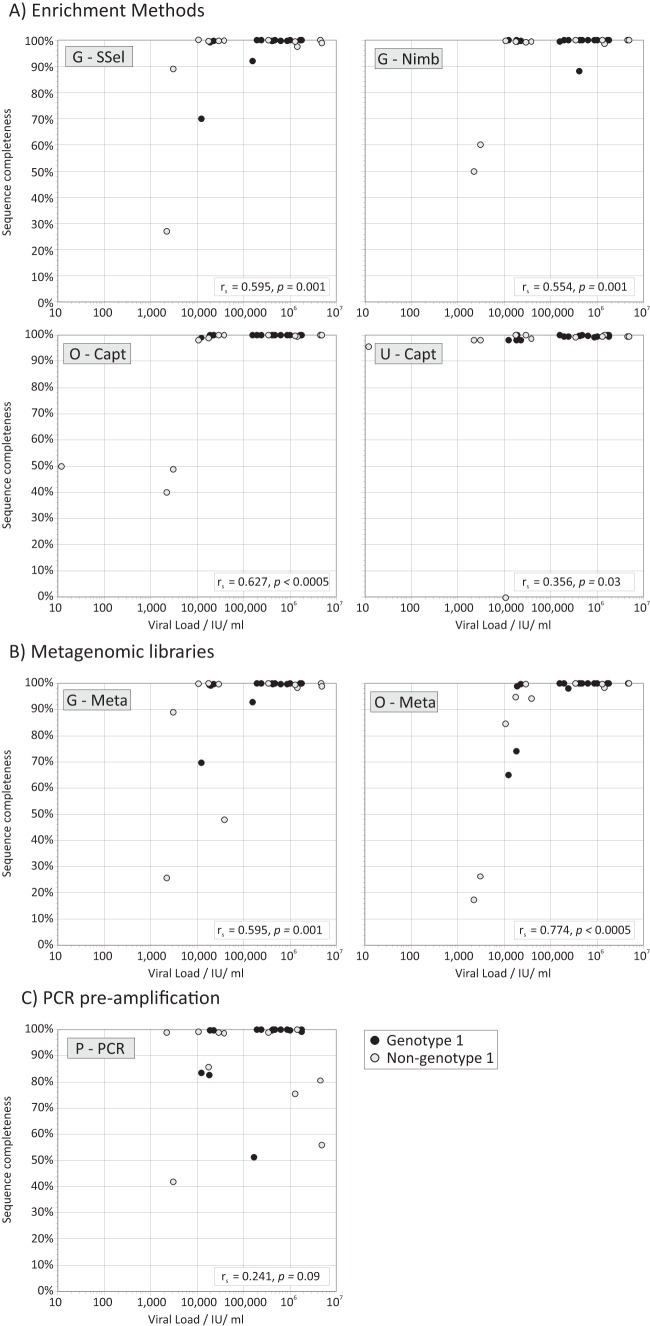
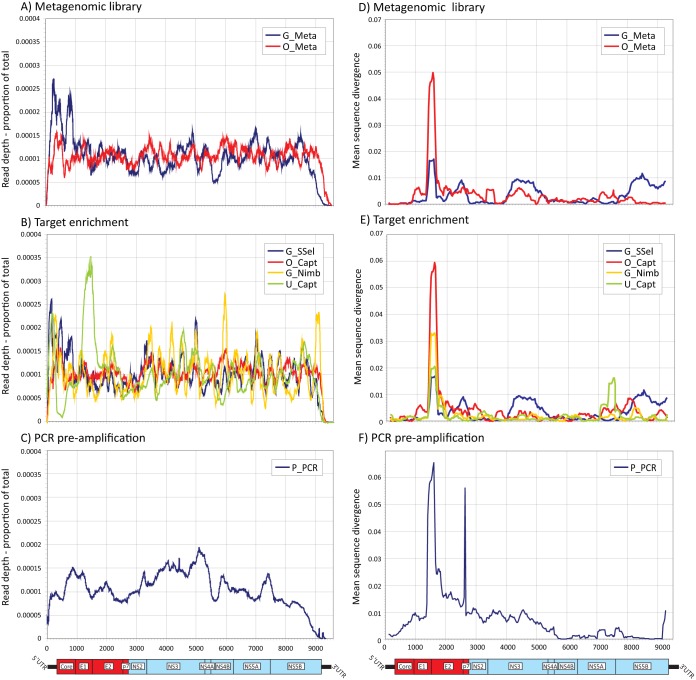
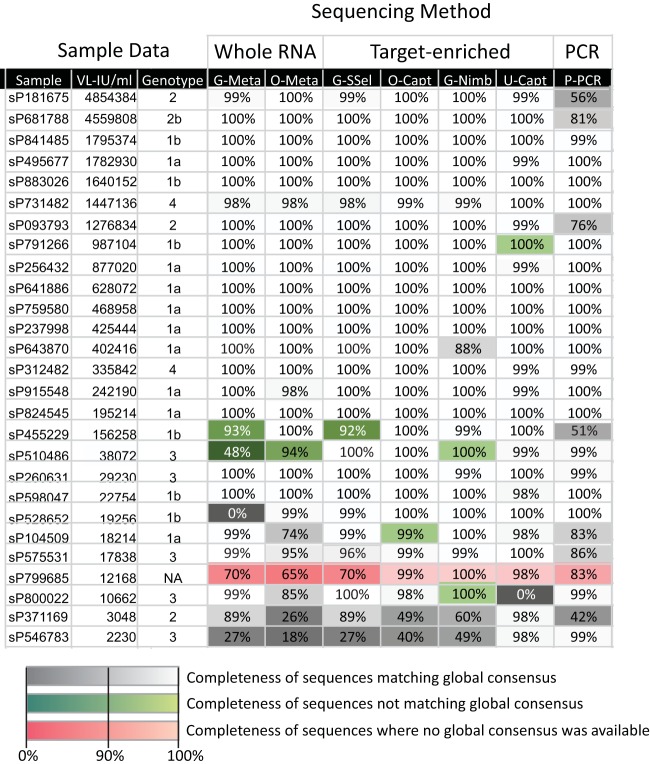
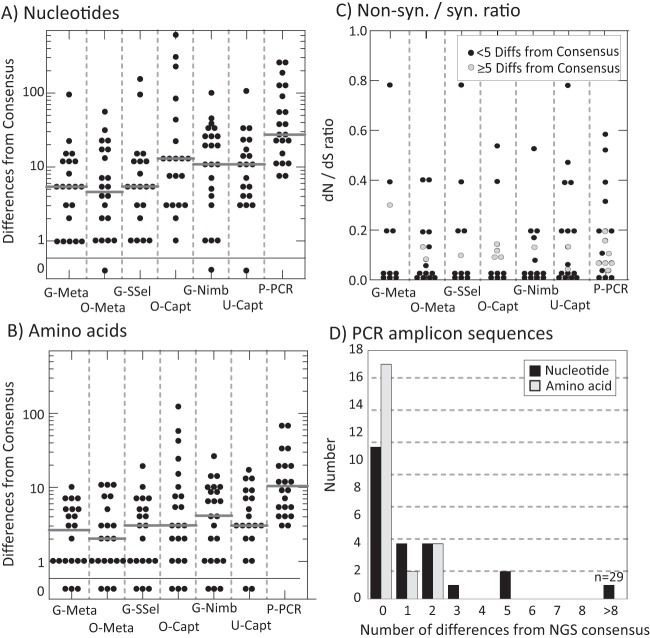
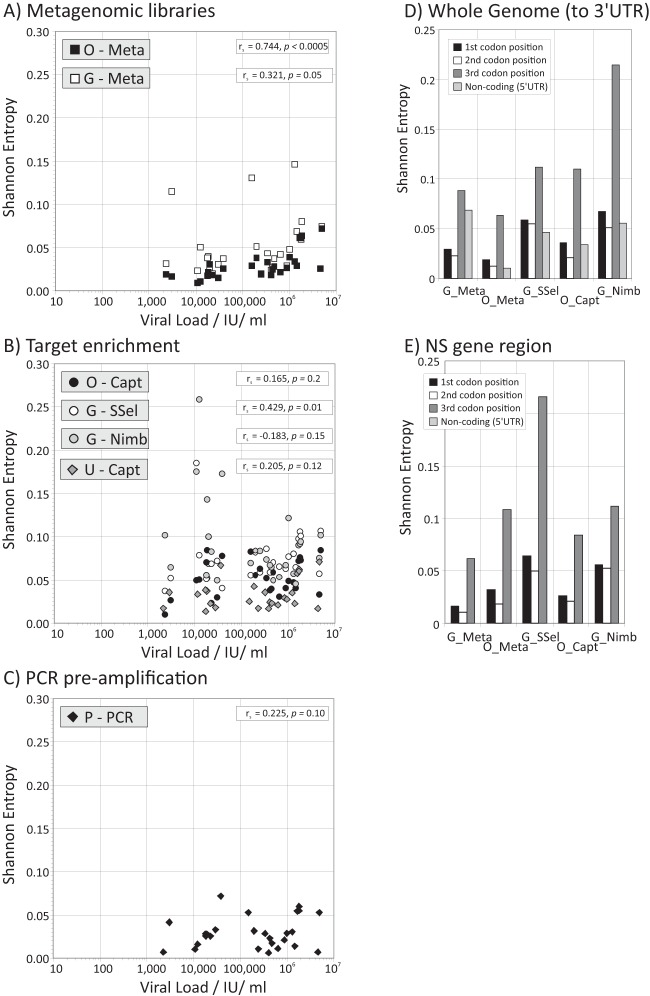
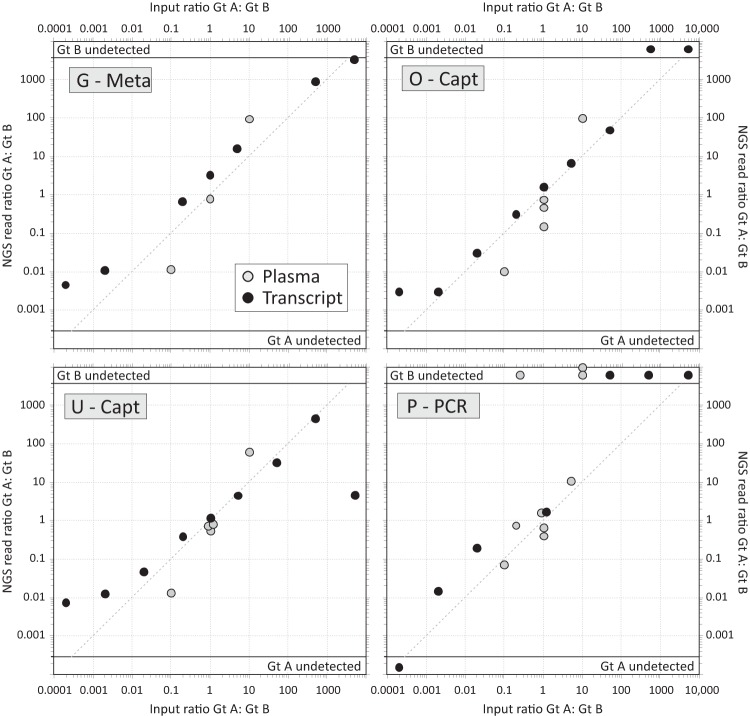
Affordable next-generation sequencing (NGS) technologies for hepatitis C virus (HCV) may potentially identify both viral genotype and resistance genetic motifs in the era of directly acting antiviral (DAA) therapies. This study compared the ability of high-throughput NGS methods to generate full-length, deep, HCV sequence data sets and evaluated their utility for diagnostics and clinical assessment. NGS methods using (i) unselected HCV RNA (metagenomics), (ii) preenrichment of HCV RNA by probe capture, and (iii) HCV preamplification by PCR implemented in four United Kingdom centers were compared. Metrics of sequence coverage and depth, quasispecies diversity, and detection of DAA resistance-associated variants (RAVs), mixed HCV genotypes, and other coinfections were compared using a panel of samples with different viral loads, genotypes, and mixed HCV genotypes/subtypes [geno(sub)types]. Each NGS method generated near-complete genome sequences from more than 90% of samples. Enrichment methods and PCR preamplification generated greater sequence depth and were more effective for samples with low viral loads. All NGS methodologies accurately identified mixed HCV genotype infections. Consensus sequences generated by different NGS methods were generally concordant, and majority RAVs were consistently detected. However, methods differed in their ability to detect minor populations of RAVs. Metagenomic methods identified human pegivirus coinfections. NGS provided a rapid, inexpensive method for generating whole HCV genomes to define infecting genotypes, RAVs, comprehensive viral strain analysis, and quasispecies diversity. Enrichment methods are particularly suited for high-throughput analysis while providing the genotype and information on potential DAA resistance.
在直接作用抗病毒药物(DAA)治疗时代,用于丙型肝炎病毒(HCV)的经济实惠的下一代测序(NGS)技术可能潜在地识别病毒基因型和耐药基因基序。本研究比较了高通量NGS方法生成全长、深度HCV序列数据集的能力,并评估了它们在诊断和临床评估中的效用。比较了在英国四个中心实施的使用(i)未选择的HCV RNA(宏基因组学)、(ii)通过探针捕获对HCV RNA进行预富集以及(iii)通过PCR进行HCV预扩增的NGS方法。使用一组具有不同病毒载量、基因型以及混合HCV基因型/亚型[基因型(亚)型]的样本,比较了序列覆盖度和深度、准种多样性以及DAA耐药相关变异(RAV)、混合HCV基因型和其他合并感染的检测指标。每种NGS方法都从超过90%的样本中生成了近乎完整的基因组序列。富集方法和PCR预扩增产生了更高的序列深度,并且对低病毒载量的样本更有效。所有NGS方法都能准确识别混合HCV基因型感染。不同NGS方法生成的共识序列通常一致,并且多数RAV被一致检测到。然而,不同方法在检测RAV少数群体的能力上存在差异。宏基因组学方法识别出了人pegivirus合并感染。NGS为生成完整HCV基因组以确定感染基因型、RAV、全面的病毒株分析和准种多样性提供了一种快速、廉价的方法。富集方法特别适合高通量分析,同时提供基因型和潜在DAA耐药性的信息。