Mohamadi Hamid, Chu Justin, Vandervalk Benjamin P, Birol Inanc
Canada's Michael Smith Genome Sciences Centre, British Columbia Cancer Agency, Vancouver, BC V5Z 4S6, Canada.
Bioinformatics. 2016 Nov 15;32(22):3492-3494. doi: 10.1093/bioinformatics/btw397. Epub 2016 Jul 16.
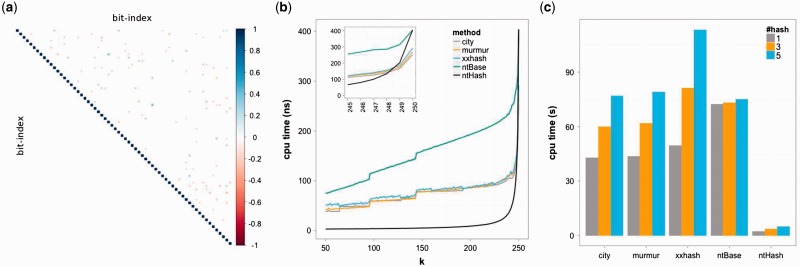
Hashing has been widely used for indexing, querying and rapid similarity search in many bioinformatics applications, including sequence alignment, genome and transcriptome assembly, k-mer counting and error correction. Hence, expediting hashing operations would have a substantial impact in the field, making bioinformatics applications faster and more efficient.
We present ntHash, a hashing algorithm tuned for processing DNA/RNA sequences. It performs the best when calculating hash values for adjacent k-mers in an input sequence, operating an order of magnitude faster than the best performing alternatives in typical use cases.
ntHash is available online at http://www.bcgsc.ca/platform/bioinfo/software/nthash and is free for academic use.
hmohamadi@bcgsc.ca or ibirol@bcgsc.caSupplementary information: Supplementary data are available at Bioinformatics online.
哈希已广泛应用于许多生物信息学应用中的索引、查询和快速相似性搜索,包括序列比对、基因组和转录组组装、k-mer计数和错误校正。因此,加速哈希运算将对该领域产生重大影响,使生物信息学应用更快、更高效。
我们提出了ntHash,一种针对处理DNA/RNA序列进行优化的哈希算法。在为输入序列中的相邻k-mer计算哈希值时,它表现最佳,在典型用例中比性能最佳的替代方案快一个数量级。
ntHash可在http://www.bcgsc.ca/platform/bioinfo/software/nthash在线获取,供学术使用免费。
hmohamadi@bcgsc.ca或ibirol@bcgsc.ca
补充数据可在《生物信息学》在线获取。