Watson Amanda J, Hopkins Gemma V, Hitchin Samantha, Begum Habiba, Jones Stuart, Jordan Allan, Holt Sarah, March H Nikki, Newton Rebecca, Small Helen, Stowell Alex, Waddell Ian D, Waszkowycz Bohdan, Ogilvie Donald J
Cancer Research UK Manchester Institute, Drug Discovery Unit, University of Manchester, Manchester, M20 4BX, UK.
F1000Res. 2016 May 26;5:1005. doi: 10.12688/f1000research.8724.2. eCollection 2016.
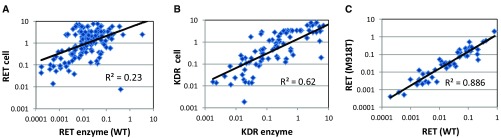
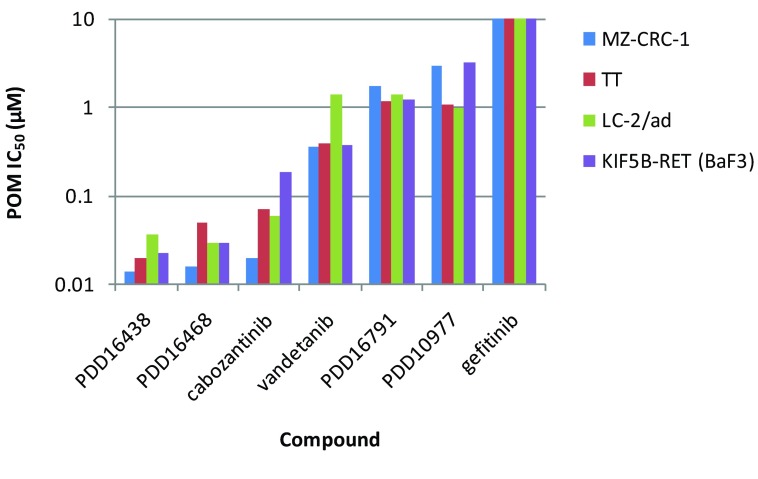
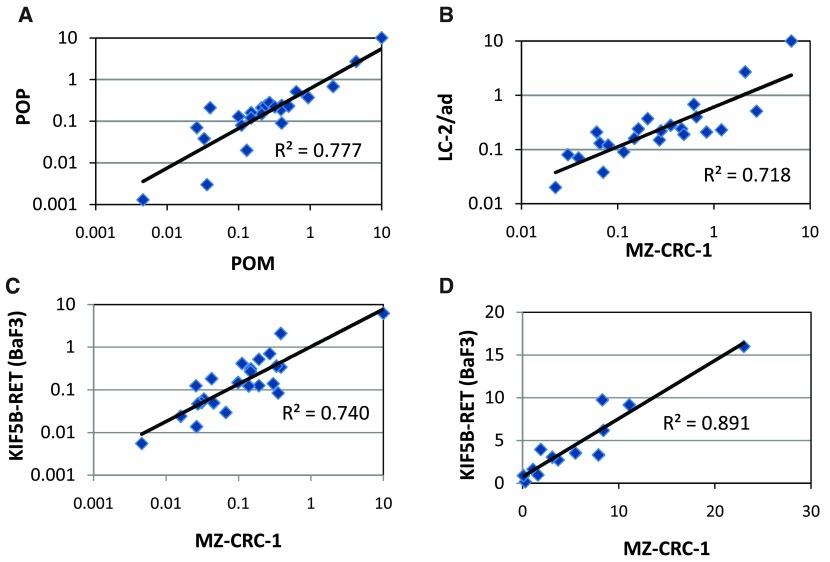
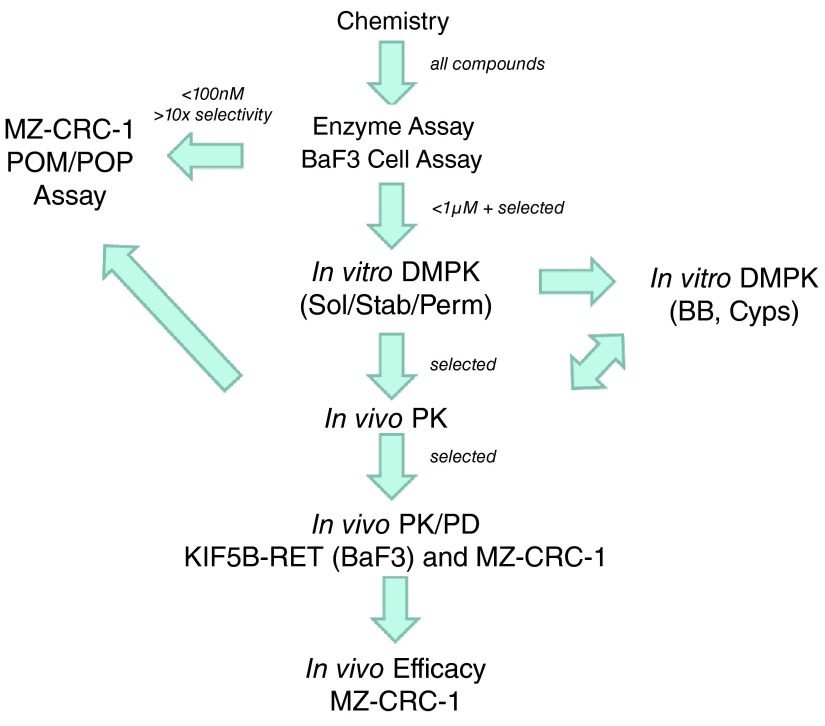
RET (REarranged during Transfection) is a receptor tyrosine kinase, which plays pivotal roles in regulating cell survival, differentiation, proliferation, migration and chemotaxis. Activation of RET is a mechanism of oncogenesis in medullary thyroid carcinomas where both germline and sporadic activating somatic mutations are prevalent. At present, there are no known specific RET inhibitors in clinical development, although many potent inhibitors of RET have been opportunistically identified through selectivity profiling of compounds initially designed to target other tyrosine kinases. Vandetanib and cabozantinib, both multi-kinase inhibitors with RET activity, are approved for use in medullary thyroid carcinoma, but additional pharmacological activities, most notably inhibition of vascular endothelial growth factor - VEGFR2 (KDR), lead to dose-limiting toxicity. The recent identification of RET fusions present in ~1% of lung adenocarcinoma patients has renewed interest in the identification and development of more selective RET inhibitors lacking the toxicities associated with the current treatments. In an earlier publication [Newton et al, 2016; 1] we reported the discovery of a series of 2-substituted phenol quinazolines as potent and selective RET kinase inhibitors. Here we describe the development of the robust screening cascade which allowed the identification and advancement of this chemical series. Furthermore we have profiled a panel of RET-active clinical compounds both to validate the cascade and to confirm that none display a RET-selective target profile.
RET(转染期间重排)是一种受体酪氨酸激酶,在调节细胞存活、分化、增殖、迁移和趋化性方面发挥着关键作用。RET的激活是甲状腺髓样癌发生肿瘤的一种机制,其中种系和散发性激活体细胞突变都很常见。目前,尚无已知的特异性RET抑制剂处于临床开发阶段,尽管通过对最初设计用于靶向其他酪氨酸激酶的化合物进行选择性分析,已偶然发现了许多有效的RET抑制剂。凡德他尼和卡博替尼都是具有RET活性的多激酶抑制剂,已被批准用于甲状腺髓样癌,但其他药理活性,最显著的是抑制血管内皮生长因子-VEGFR2(KDR),会导致剂量限制性毒性。最近在约1%的肺腺癌患者中发现了RET融合,这重新激发了人们对鉴定和开发更具选择性、且无当前治疗相关毒性的RET抑制剂的兴趣。在早期的一篇出版物中[牛顿等人,2016年;1],我们报告了一系列2-取代苯酚喹唑啉作为强效和选择性RET激酶抑制剂的发现。在此,我们描述了强大的筛选级联反应的开发过程,该过程使得能够鉴定和推进这个化学系列。此外,我们对一组具有RET活性的临床化合物进行了分析,以验证该级联反应,并确认没有一种化合物显示出RET选择性靶点特征。