Friedrich Thomas, Tavraz Neslihan N, Junghans Cornelia
Department of Physical Chemistry/Bioenergetics, Institute of Chemistry, Technical University of Berlin Berlin, Germany.
Front Physiol. 2016 Jun 21;7:239. doi: 10.3389/fphys.2016.00239. eCollection 2016.
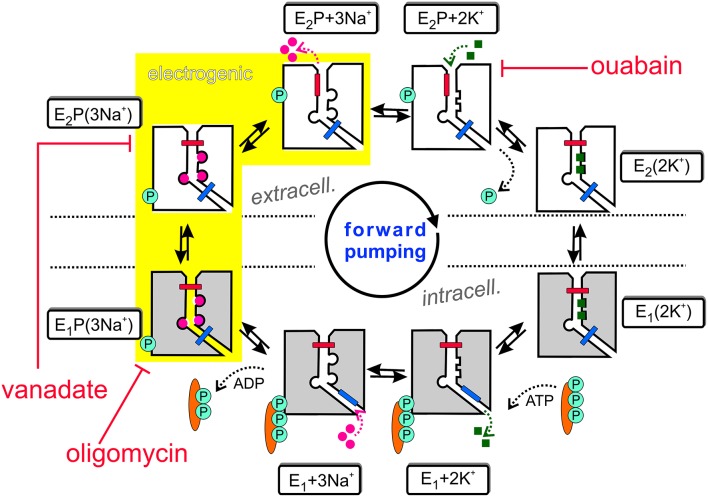
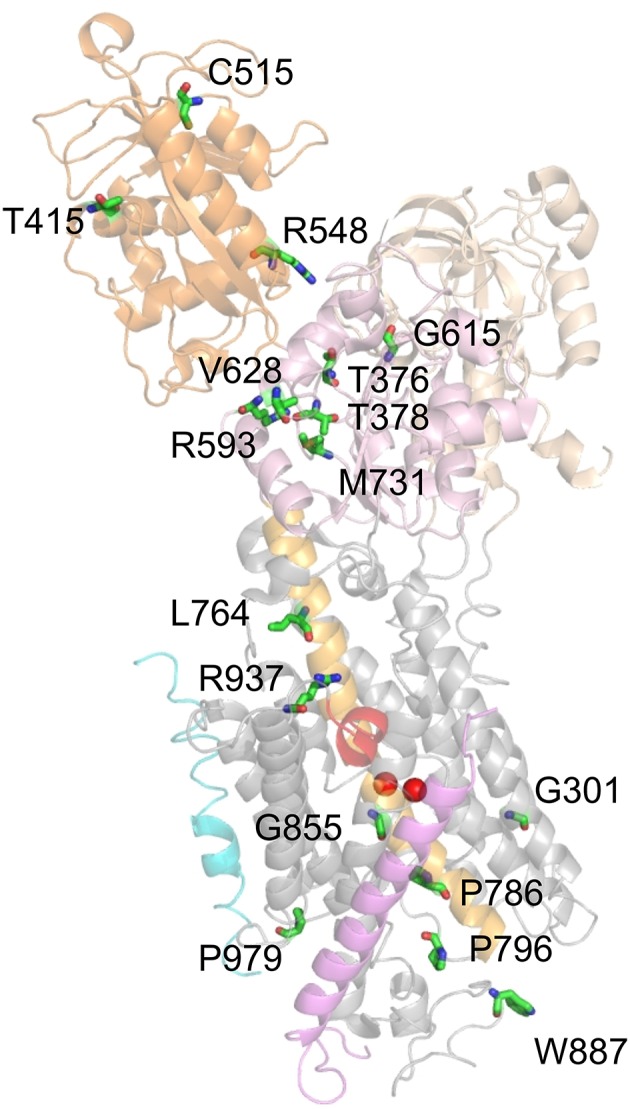
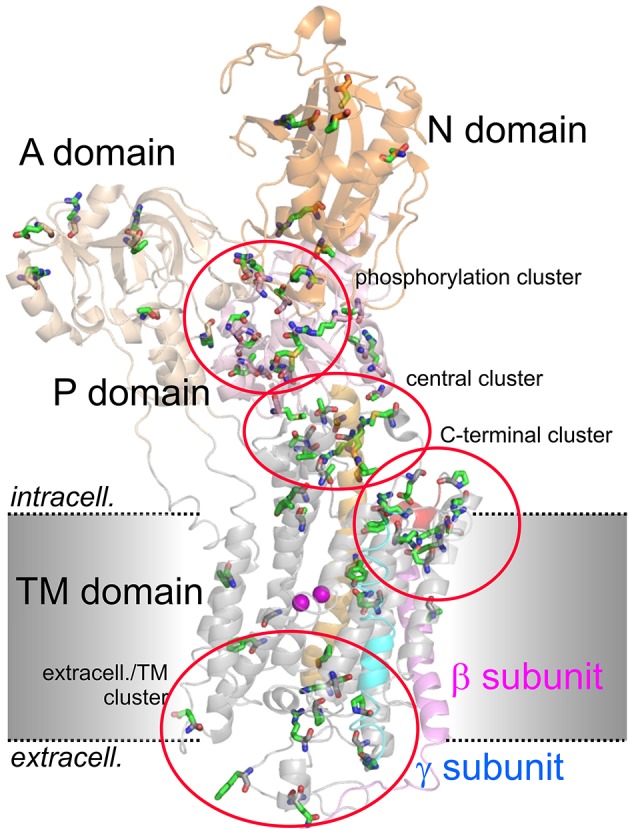
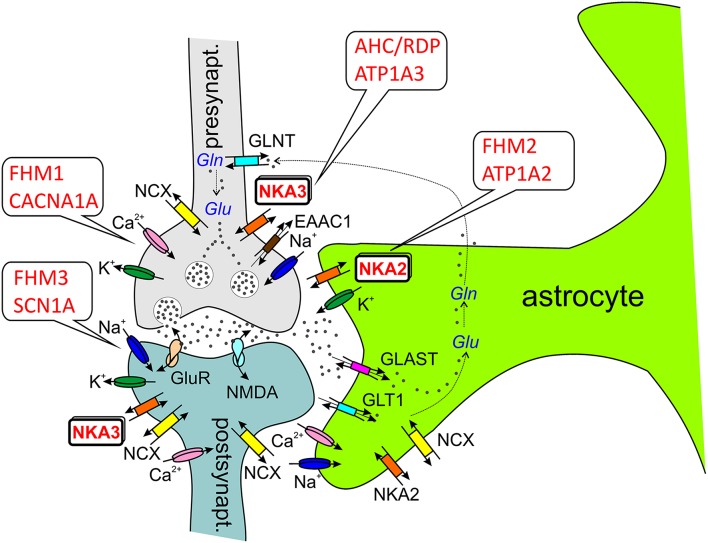
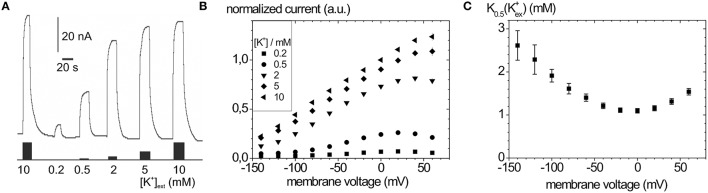
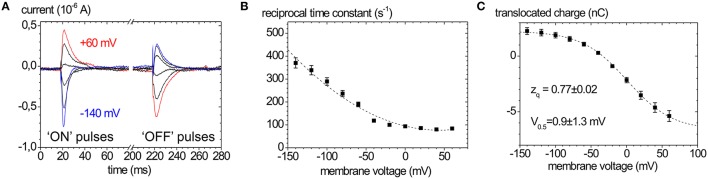
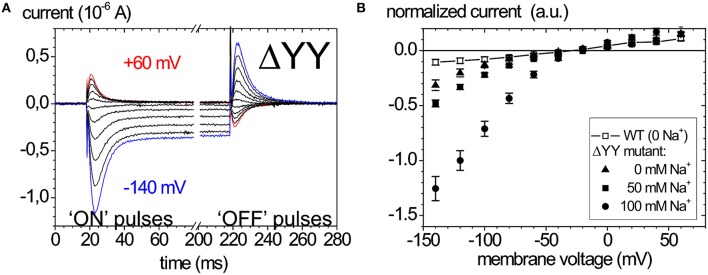
Mutations in four genes have been identified in familial hemiplegic migraine (FHM), from which CACNA1A (FHM type 1) and SCN1A (FHM type 3) code for neuronal voltage-gated calcium or sodium channels, respectively, while ATP1A2 (FHM type 2) encodes the α2 isoform of the Na(+),K(+)-ATPase's catalytic subunit, thus classifying FHM primarily as an ion channel/ion transporter pathology. FHM type 4 is attributed to mutations in the PRRT2 gene, which encodes a proline-rich transmembrane protein of as yet unknown function. The Na(+),K(+)-ATPase maintains the physiological gradients for Na(+) and K(+) ions and is, therefore, critical for the activity of ion channels and transporters involved neuronal excitability, neurotransmitter uptake or Ca(2+) signaling. Strikingly diverse functional abnormalities have been identified for disease-linked ATP1A2 mutations which frequently lead to changes in the enzyme's voltage-dependent properties, kinetics, or apparent cation affinities, but some mutations are truly deleterious for enzyme function and thus cause full haploinsufficiency. Here, we summarize structural and functional data about the Na(+),K(+)-ATPase available to date and an overview is provided about the particular properties of the α2 isoform that explain its physiological relevance in electrically excitable tissues. In addition, current concepts about the neurobiology of migraine, the correlations between primary brain dysfunction and mechanisms of headache pain generation are described, together with insights gained recently from modeling approaches in computational neuroscience. Then, a survey is given about ATP1A2 mutations implicated in migraine cases as documented in the literature with focus on mutations that were described to completely destroy enzyme function, or lead to misfolded or mistargeted protein in particular model cell lines. We also discuss whether or not there are correlations between these most severe mutational effects and clinical phenotypes. Finally, perspectives for future research on the implications of Na(+),K(+)-ATPase mutations in human pathologies are presented.
在家族性偏瘫性偏头痛(FHM)中已鉴定出四个基因的突变,其中CACNA1A(FHM 1型)和SCN1A(FHM 3型)分别编码神经元电压门控钙通道或钠通道,而ATP1A2(FHM 2型)编码Na⁺,K⁺-ATP酶催化亚基的α2同工型,因此FHM主要被归类为离子通道/离子转运体病理学。FHM 4型归因于PRRT2基因的突变,该基因编码一种功能尚不清楚的富含脯氨酸的跨膜蛋白。Na⁺,K⁺-ATP酶维持Na⁺和K⁺离子的生理梯度,因此对于参与神经元兴奋性、神经递质摄取或Ca²⁺信号传导的离子通道和转运体的活性至关重要。已发现与疾病相关的ATP1A2突变存在惊人的多种功能异常,这些突变经常导致酶的电压依赖性特性、动力学或明显的阳离子亲和力发生变化,但有些突变对酶功能确实有害,从而导致完全的单倍体不足。在这里,我们总结了迄今为止关于Na⁺,K⁺-ATP酶的结构和功能数据,并概述了α2同工型的特殊性质,这些性质解释了其在电可兴奋组织中的生理相关性。此外,还描述了当前关于偏头痛神经生物学的概念、原发性脑功能障碍与头痛疼痛产生机制之间的相关性,以及最近从计算神经科学建模方法中获得的见解。然后,对文献中记录的与偏头痛病例相关的ATP1A2突变进行了综述,重点关注那些被描述为完全破坏酶功能或导致特定模型细胞系中蛋白质错误折叠或靶向错误的突变。我们还讨论了这些最严重的突变效应与临床表型之间是否存在相关性。最后,提出了关于Na⁺,K⁺-ATP酶突变在人类病理学中的影响的未来研究展望。