Rush Eric T, Hartmann Julianne E, Skrabal Jill C, Rizzo William B
Munroe-Meyer Institute for Genetics and Rehabilitation, University of Nebraska Medical Center, Omaha, NE, USA; Department of Pediatrics, University of Nebraska Medical Center, Omaha, NE, USA.
Munroe-Meyer Institute for Genetics and Rehabilitation, University of Nebraska Medical Center, Omaha, NE, USA.
SAGE Open Med Case Rep. 2014 Jul 31;2:2050313X14546348. doi: 10.1177/2050313X14546348. eCollection 2014.
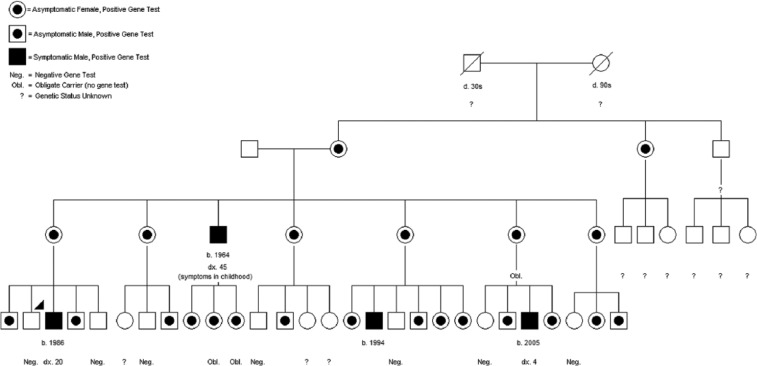
Ornithine transcarbamylase deficiency is the most common inherited disorder of the urea cycle, has a variable phenotype, and is caused by mutations in the OTC gene. We report three cases of ornithine transcarbamylase deficiency to illustrate the late-onset presentation of this disorder and provide strategies for diagnosis and treatment. The patients were maternal first cousins, presenting with hyperammonemia and obtundation. Urea cycle disorder was not initially suspected in the first patient, delaying diagnosis.
Sequencing of the OTC gene showed a novel missense mutation, c.563G > C (p.G188A). Numerous family members were found to carry this mutation, which shows a trend toward later onset. Each urea cycle disorder has its own unique pattern of biochemical abnormalities, which differ from non-metabolic causes of critical illness.
Regardless of age, clinical suspicion of a urea cycle disorder is important in encephalopathic patients to ensure quick diagnosis and definitive treatment of the underlying inborn error of metabolism.
鸟氨酸转氨甲酰酶缺乏症是尿素循环中最常见的遗传性疾病,具有可变的表型,由OTC基因突变引起。我们报告三例鸟氨酸转氨甲酰酶缺乏症病例,以说明该疾病的迟发性表现,并提供诊断和治疗策略。这些患者是母系第一代堂表亲,表现为高氨血症和意识不清。首例患者最初未怀疑尿素循环障碍,导致诊断延迟。
OTC基因测序显示一个新的错义突变,c.563G>C(p.G188A)。发现许多家庭成员携带此突变,显示出发病较晚的趋势。每种尿素循环障碍都有其独特的生化异常模式,这与危重病的非代谢原因不同。
无论年龄大小,对于脑病患者,临床怀疑尿素循环障碍对于确保快速诊断和明确治疗潜在的先天性代谢缺陷很重要。