Satoh Takashi P, Miya Masaki, Mabuchi Kohji, Nishida Mutsumi
Atmosphere and Ocean Research Institute, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa City, Chiba, 277-8654, Japan.
Collection Center, National Museum of Nature and Science, 4-1-1 Amakubo, Tsukuba City, Ibaraki, 305-0005, Japan.
BMC Genomics. 2016 Sep 7;17(1):719. doi: 10.1186/s12864-016-3054-y.
The mitochondrial (mt) genome has been used as an effective tool for phylogenetic and population genetic analyses in vertebrates. However, the structure and variability of the vertebrate mt genome are not well understood. A potential strategy for improving our understanding is to conduct a comprehensive comparative study of large mt genome data. The aim of this study was to characterize the structure and variability of the fish mt genome through comparative analysis of large datasets.
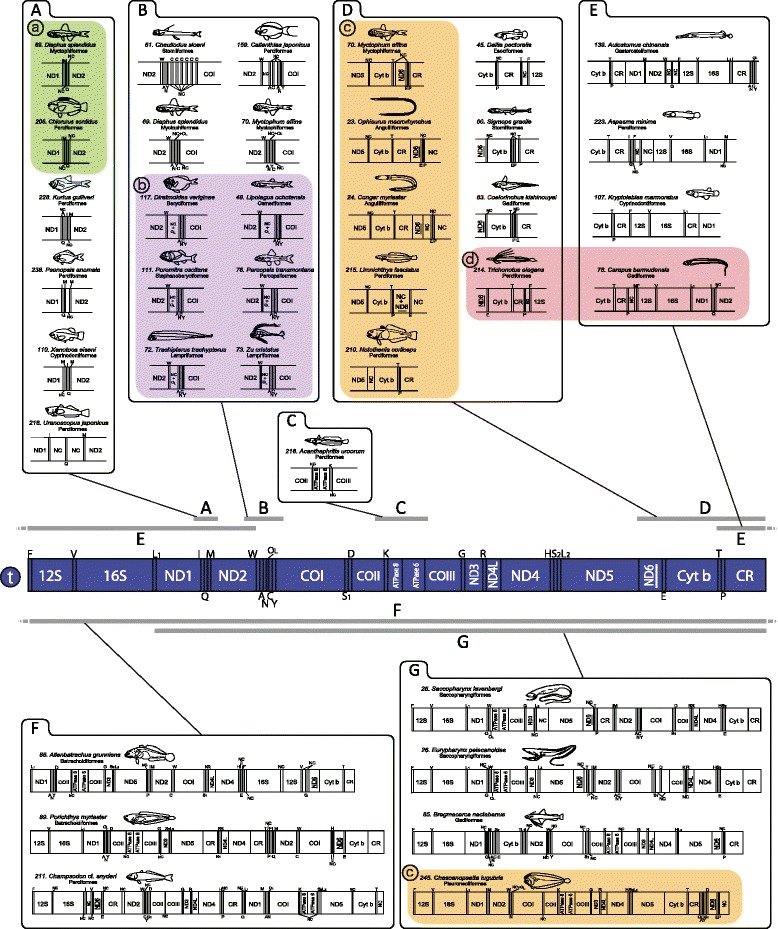
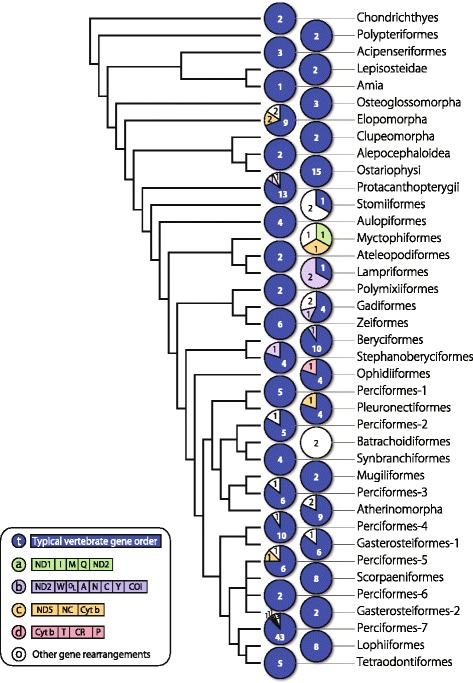
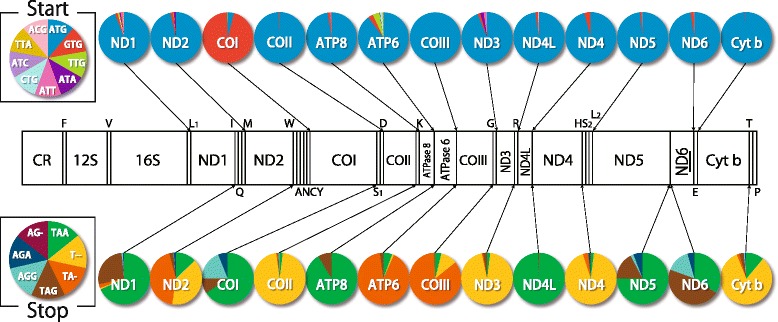
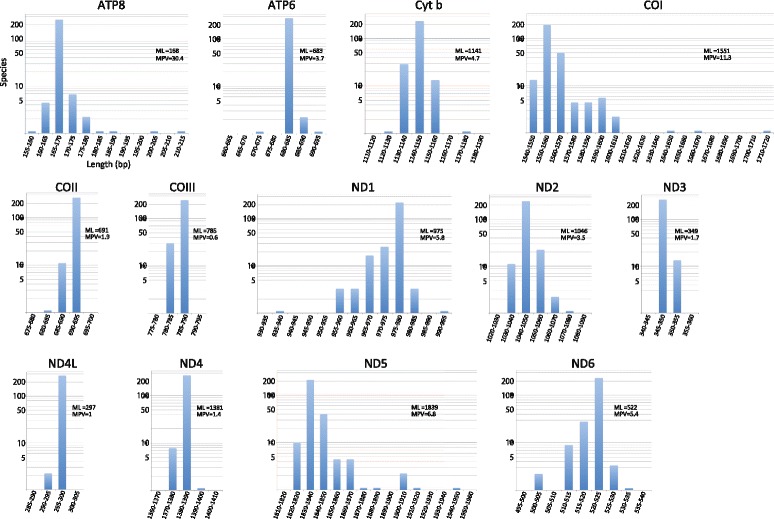
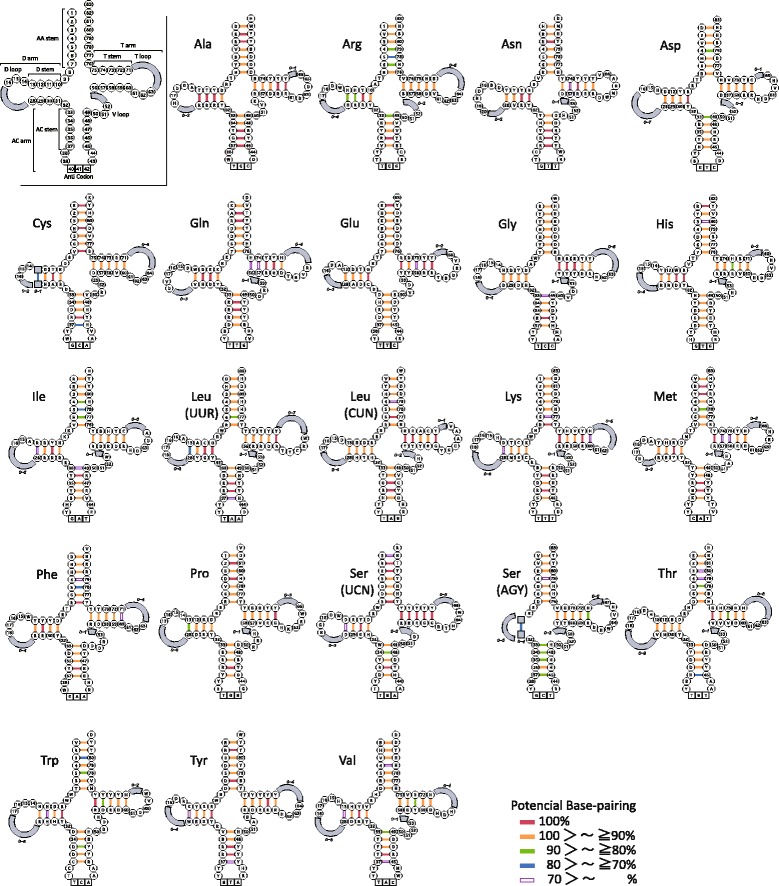
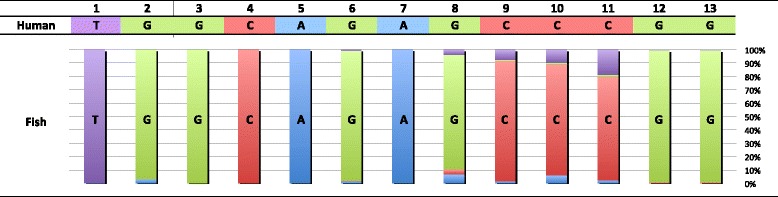
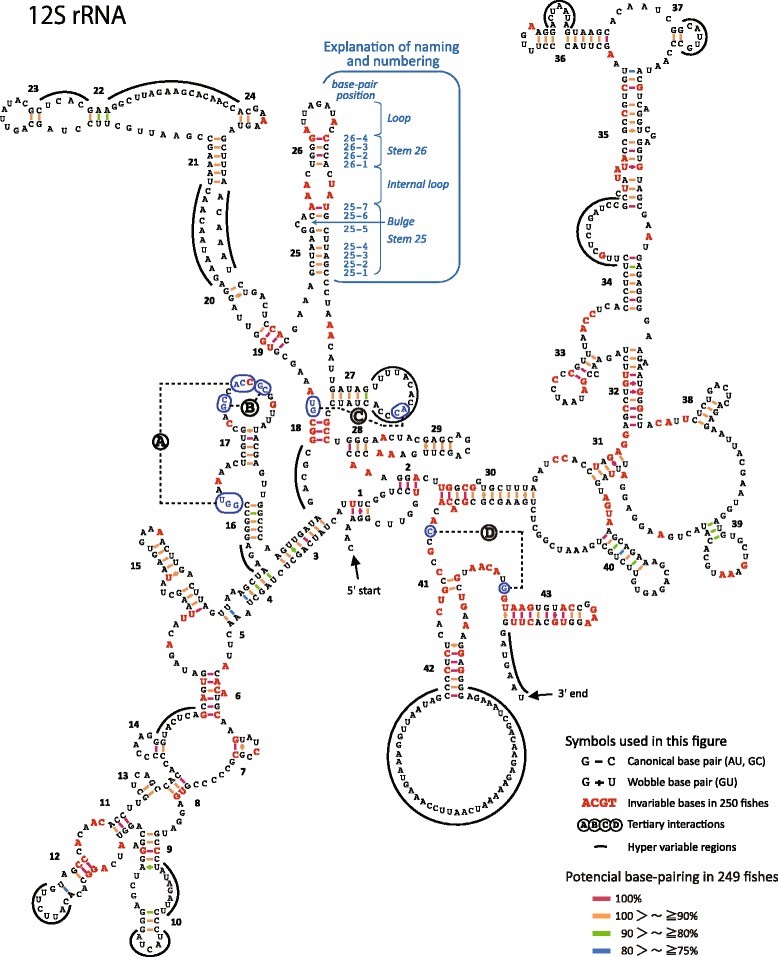
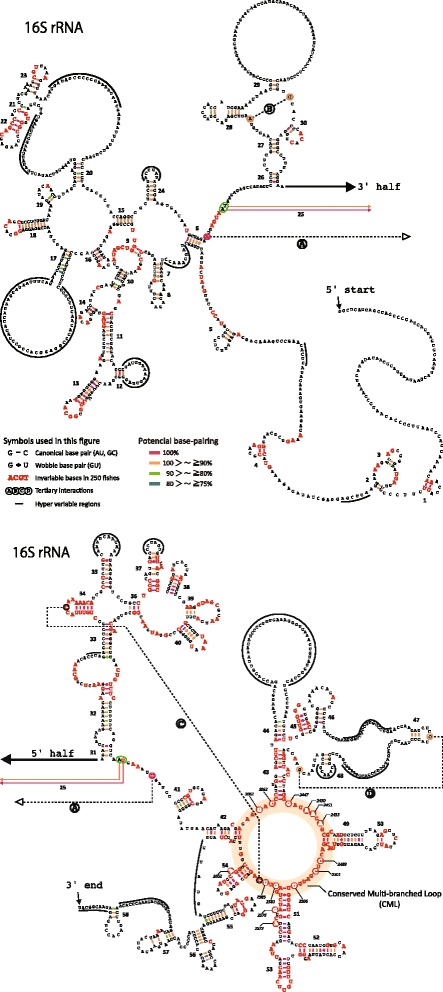
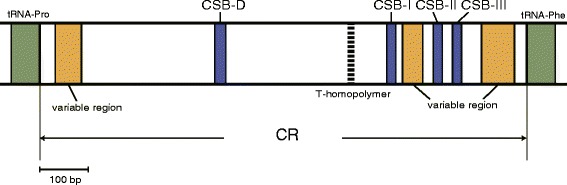
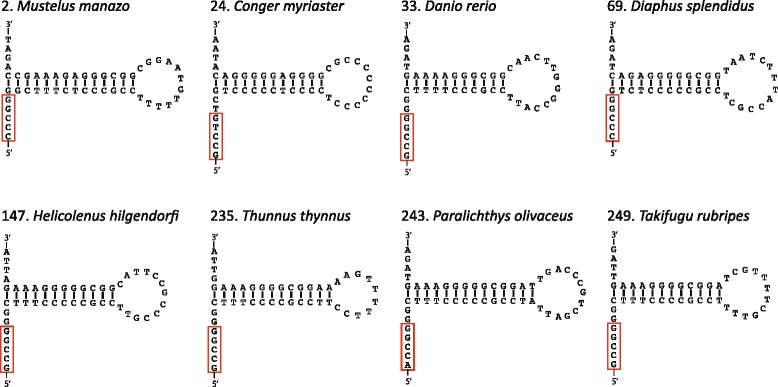
An analysis of the secondary structure of proteins for 250 fish species (248 ray-finned and 2 cartilaginous fishes) illustrated that cytochrome c oxidase subunits (COI, COII, and COIII) and a cytochrome bc1 complex subunit (Cyt b) had substantial amino acid conservation. Among the four proteins, COI was the most conserved, as more than half of all amino acid sites were invariable among the 250 species. Our models identified 43 and 58 stems within 12S rRNA and 16S rRNA, respectively, with larger numbers than proposed previously for vertebrates. The models also identified 149 and 319 invariable sites in 12S rRNA and 16S rRNA, respectively, in all fishes. In particular, the present result verified that a region corresponding to the peptidyl transferase center in prokaryotic 23S rRNA, which is homologous to mt 16S rRNA, is also conserved in fish mt 16S rRNA. Concerning the gene order, we found 35 variations (in 32 families) that deviated from the common gene order in vertebrates. These gene rearrangements were mostly observed in the area spanning the ND5 gene to the control region as well as two tRNA gene cluster regions (IQM and WANCY regions). Although many of such gene rearrangements were unique to a specific taxon, some were shared polyphyletically between distantly related species.
Through a large-scale comparative analysis of 250 fish species mt genomes, we elucidated various structural aspects of the fish mt genome and the encoded genes. The present results will be important for understanding functions of the mt genome and developing programs for nucleotide sequence analysis. This study demonstrated the significance of extensive comparisons for understanding the structure of the mt genome.
线粒体(mt)基因组已被用作脊椎动物系统发育和群体遗传学分析的有效工具。然而,脊椎动物mt基因组的结构和变异性尚未得到充分了解。提高我们理解的一个潜在策略是对大量mt基因组数据进行全面的比较研究。本研究的目的是通过对大量数据集的比较分析来表征鱼类mt基因组的结构和变异性。
对250种鱼类(248种硬骨鱼和2种软骨鱼)蛋白质二级结构的分析表明,细胞色素c氧化酶亚基(COI、COII和COIII)和细胞色素bc1复合体亚基(Cyt b)具有显著的氨基酸保守性。在这四种蛋白质中,COI最保守,因为在250个物种中超过一半的氨基酸位点是不变的。我们的模型分别在12S rRNA和16S rRNA中鉴定出43个和58个茎,数量比之前对脊椎动物的提议更多。这些模型还分别在所有鱼类的12S rRNA和16S rRNA中鉴定出149个和319个不变位点。特别是,目前的结果证实,与mt 16S rRNA同源的原核生物2 s rRNA中与肽基转移酶中心相对应的区域在鱼类mt 16S rRNA中也保守。关于基因顺序,我们发现35种变异(在32个科中)偏离了脊椎动物的常见基因顺序。这些基因重排大多出现在从ND5基因到控制区以及两个tRNA基因簇区域(IQM和WANCY区域)的区域。虽然许多这样的基因重排是特定分类群特有的,但有些是远缘物种之间多系共享的。
通过对250种鱼类mt基因组的大规模比较分析,我们阐明了鱼类mt基因组和编码基因的各种结构方面。目前的结果对于理解mt基因组的功能和开发核苷酸序列分析程序将是重要的。本研究证明了广泛比较对于理解mt基因组结构的重要性。