Linhares Natália D, Valadares Eugênia R, da Costa Silvia S, Arantes Rodrigo R, de Oliveira Luiz Roberto, Rosenberg Carla, Vianna-Morgante Angela M, Svartman Marta
Setor de Citogenética/Laboratório Central do Hospital das Clínicas da Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
Departamento de Propedêutica Complementar, Faculdade de Medicina, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil; Ambulatório de Erros Inatos do Metabolismo, Hospital das Clínicas da Universidade Federal de Minas Gerais, Belo Horizonte, Brazil.
Meta Gene. 2016 Jul 7;9:185-90. doi: 10.1016/j.mgene.2016.07.004. eCollection 2016 Sep.
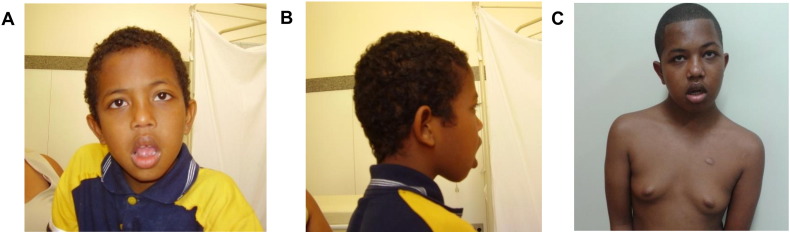
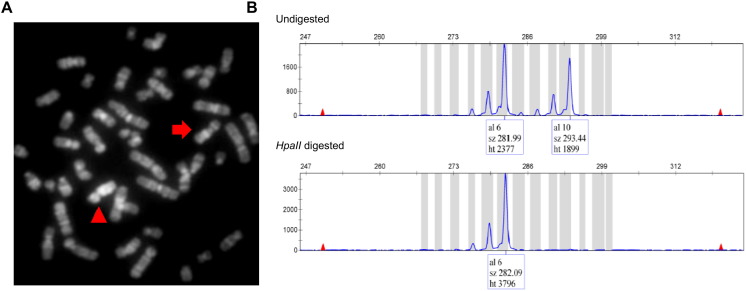
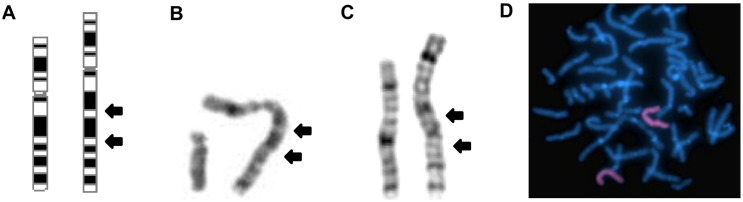
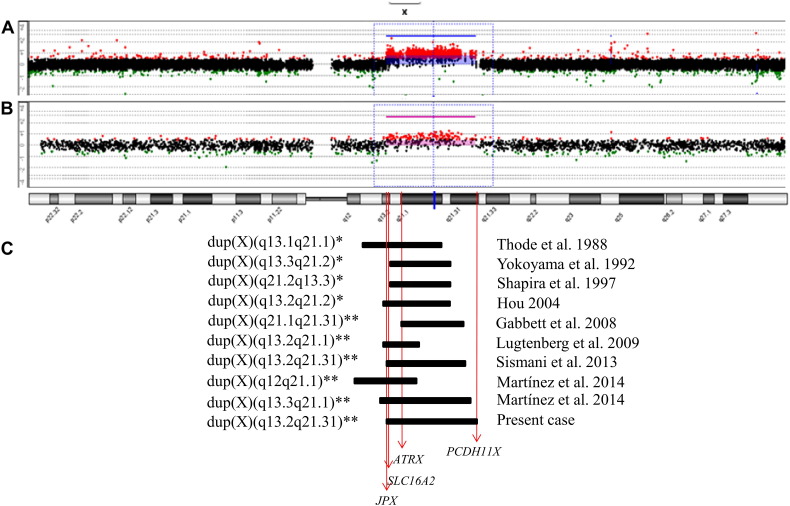
We report on a 16-year-old boy with a maternally inherited ~ 18.3 Mb Xq13.2-q21.31 duplication delimited by aCGH. As previously described in patients with similar duplications, his clinical features included intellectual disability, developmental delay, speech delay, generalized hypotonia, infantile feeding difficulties, self-injurious behavior, short stature and endocrine problems. As additional findings, he presented recurrent seizures and pubertal gynecomastia. His mother was phenotypically normal and had completely skewed inactivation of the duplicated X chromosome, as most female carriers of such duplications. Five previously reported patients with partial Xq duplications presented duplication breakpoints similar to those of our patient. One of them, a fetus with multiple congenital abnormalities, had the same cytogenetic duplication breakpoint. Three of the reported patients shared many features with our proband but the other had some clinical features of the Prader-Willi syndrome. It was suggested that ATRX overexpression could be involved in the major clinical features of patients with partial Xq duplications. We propose that this gene could also be involved with the obesity of the patient with the Prader-Willi-like phenotype. Additionally, we suggest that the PCDH11X gene could be a candidate for our patient's recurrent seizures. In males, the Xq13-q21 duplication should be considered in the differential diagnosis of Prader-Willi syndrome, as previously suggested, and neuromuscular diseases, particularly mitochondriopathies.
我们报告了一名16岁男孩,其通过比较基因组杂交(aCGH)检测到母系遗传的约18.3 Mb Xq13.2-q21.31重复。正如先前在具有类似重复的患者中所描述的那样,他的临床特征包括智力残疾、发育迟缓、语言迟缓、全身肌张力减退、婴儿期喂养困难、自伤行为、身材矮小和内分泌问题。另外,他还出现了反复发作的癫痫和青春期乳腺增生。他的母亲表型正常,并且与大多数此类重复的女性携带者一样,其重复的X染色体完全失活。先前报道的5例部分Xq重复患者的重复断点与我们的患者相似。其中1例患有多种先天性异常的胎儿具有相同的细胞遗传学重复断点。报道的患者中有3例与我们的先证者有许多共同特征,但另1例具有普拉德-威利综合征的一些临床特征。有人提出,ATRX过表达可能与部分Xq重复患者的主要临床特征有关。我们提出该基因也可能与具有普拉德-威利样表型患者的肥胖有关。此外,我们认为PCDH11X基因可能是我们患者反复发作癫痫的一个候选基因。如先前所建议的,在男性中,Xq13-q21重复在普拉德-威利综合征以及神经肌肉疾病,尤其是线粒体疾病的鉴别诊断中应予以考虑。