Singer Esther, Andreopoulos Bill, Bowers Robert M, Lee Janey, Deshpande Shweta, Chiniquy Jennifer, Ciobanu Doina, Klenk Hans-Peter, Zane Matthew, Daum Christopher, Clum Alicia, Cheng Jan-Fang, Copeland Alex, Woyke Tanja
DOE Joint Genome Institute, Walnut Creek, California 94598, USA.
Newcastle University, Newcastle upon Tyne, NE1 7RU, UK.
Sci Data. 2016 Sep 27;3:160081. doi: 10.1038/sdata.2016.81.
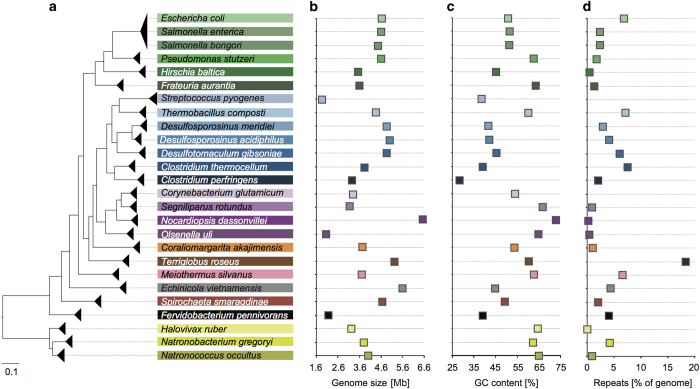
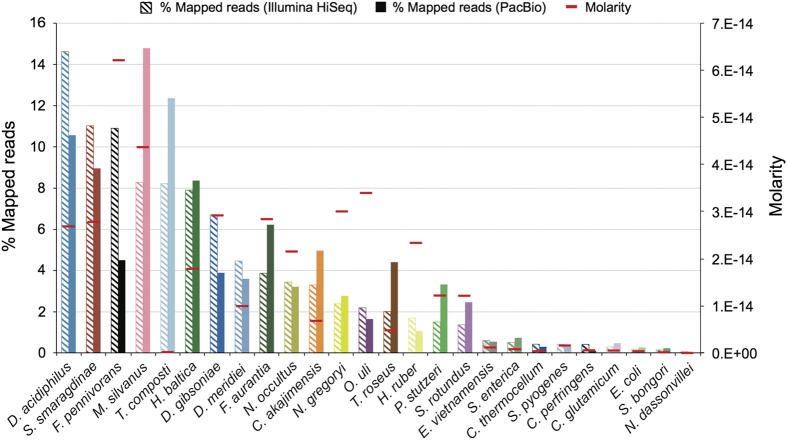
Generating sequence data of a defined community composed of organisms with complete reference genomes is indispensable for the benchmarking of new genome sequence analysis methods, including assembly and binning tools. Moreover the validation of new sequencing library protocols and platforms to assess critical components such as sequencing errors and biases relies on such datasets. We here report the next generation metagenomic sequence data of a defined mock community (Mock Bacteria ARchaea Community; MBARC-26), composed of 23 bacterial and 3 archaeal strains with finished genomes. These strains span 10 phyla and 14 classes, a range of GC contents, genome sizes, repeat content and encompass a diverse abundance profile. Short read Illumina and long-read PacBio SMRT sequences of this mock community are described. These data represent a valuable resource for the scientific community, enabling extensive benchmarking and comparative evaluation of bioinformatics tools without the need to simulate data. As such, these data can aid in improving our current sequence data analysis toolkit and spur interest in the development of new tools.
生成由具有完整参考基因组的生物体组成的特定群落的序列数据,对于包括组装和分箱工具在内的新基因组序列分析方法的基准测试而言必不可少。此外,新测序文库协议和平台的验证,以评估诸如测序错误和偏差等关键组件,也依赖于此类数据集。我们在此报告了一个特定模拟群落(模拟细菌古菌群落;MBARC - 26)的新一代宏基因组序列数据,该群落由23种细菌和3种具有完整基因组的古菌菌株组成。这些菌株涵盖10个门和14个纲,具有一系列的GC含量、基因组大小、重复含量,并包含多样化的丰度分布。本文描述了该模拟群落的短读长Illumina序列和长读长PacBio SMRT序列。这些数据为科学界提供了宝贵的资源,无需模拟数据就能对生物信息学工具进行广泛的基准测试和比较评估。因此,这些数据有助于改进我们当前的序列数据分析工具包,并激发对新工具开发的兴趣。