Tsitkanou Stavroula, Della Gatta Paul A, Russell Aaron P
Athletics Laboratory, School of Physical Education and Sport Science, University of Athens Athens, Greece.
School of Exercise and Nutrition Sciences, Institute for Physical Activity and Nutrition (IPAN), Deakin University Geelong, VIC, Australia.
Front Physiol. 2016 Sep 13;7:403. doi: 10.3389/fphys.2016.00403. eCollection 2016.
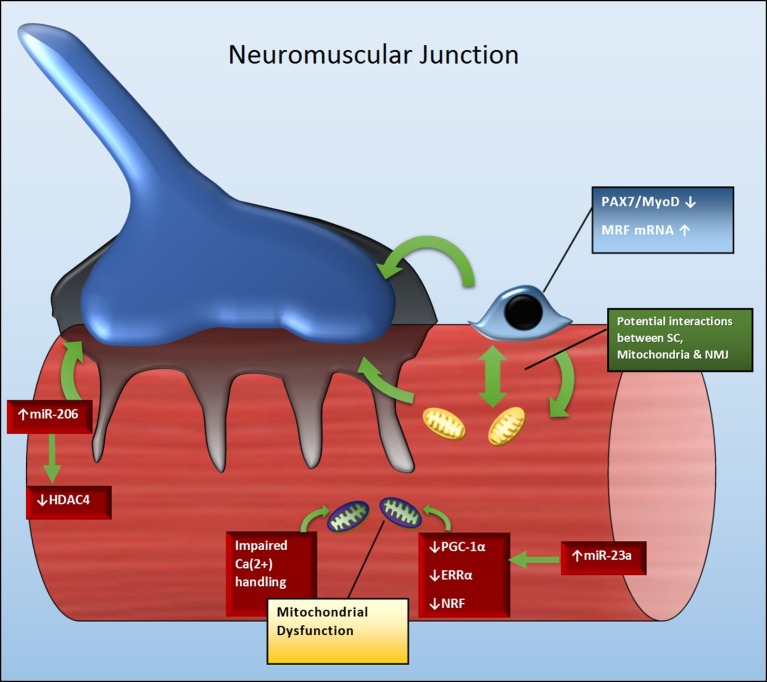
Amyotrophic lateral sclerosis (ALS), also known as motor neuron disease (MND), is a fatal motor neuron disorder. It results in progressive degeneration and death of upper and lower motor neurons, protein aggregation, severe muscle atrophy and respiratory insufficiency. Median survival with ALS is between 2 and 5 years from the onset of symptoms. ALS manifests as either familial ALS (FALS) (10% of cases) or sporadic ALS (SALS), (90% of cases). Mutations in the copper/zinc (CuZn) superoxide dismutase (SOD1) gene account for ~20% of FALS cases and the mutant SOD1 mouse model has been used extensively to help understand the ALS pathology. As the precise mechanisms causing ALS are not well understood there is presently no cure. Recent evidence suggests that motor neuron degradation may involve a cell non-autonomous phenomenon involving numerous cell types within various tissues. Skeletal muscle is now considered as an important tissue involved in the pathogenesis of ALS by activating a retrograde signaling cascade that degrades motor neurons. Skeletal muscle heath and function are regulated by numerous factors including satellite cells, mitochondria and microRNAs. Studies demonstrate that in ALS these factors show various levels of dysregulation within the skeletal muscle. This review provides an overview of their dysregulation in various ALS models as well as how they may contribute individually and/or synergistically to the ALS pathogenesis.
肌萎缩侧索硬化症(ALS),也被称为运动神经元病(MND),是一种致命的运动神经元疾病。它会导致上下运动神经元进行性退化和死亡、蛋白质聚集、严重的肌肉萎缩以及呼吸功能不全。ALS患者从症状出现起的中位生存期为2至5年。ALS表现为家族性ALS(FALS,约占病例的10%)或散发性ALS(SALS,约占病例的90%)。铜/锌(CuZn)超氧化物歧化酶(SOD1)基因突变约占FALS病例的20%,突变型SOD1小鼠模型已被广泛用于帮助理解ALS的病理学。由于导致ALS的确切机制尚未完全清楚,目前尚无治愈方法。最近的证据表明,运动神经元退化可能涉及一种细胞非自主现象,涉及各种组织内的多种细胞类型。骨骼肌现在被认为是通过激活一种逆行信号级联反应来降解运动神经元,从而参与ALS发病机制的重要组织。骨骼肌的健康和功能受多种因素调节,包括卫星细胞、线粒体和微小RNA。研究表明,在ALS中,这些因素在骨骼肌内表现出不同程度的失调。本综述概述了它们在各种ALS模型中的失调情况,以及它们如何单独和/或协同作用于ALS的发病机制。