Shafee Thomas, Cooke Ira
Department of Biochemistry and Genetics, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, 3086, Australia.
Department of Molecular and Cell Biology, James Cook University, Townsville, 4811, Australia.
BMC Bioinformatics. 2016 Oct 26;17(1):434. doi: 10.1186/s12859-016-1300-6.
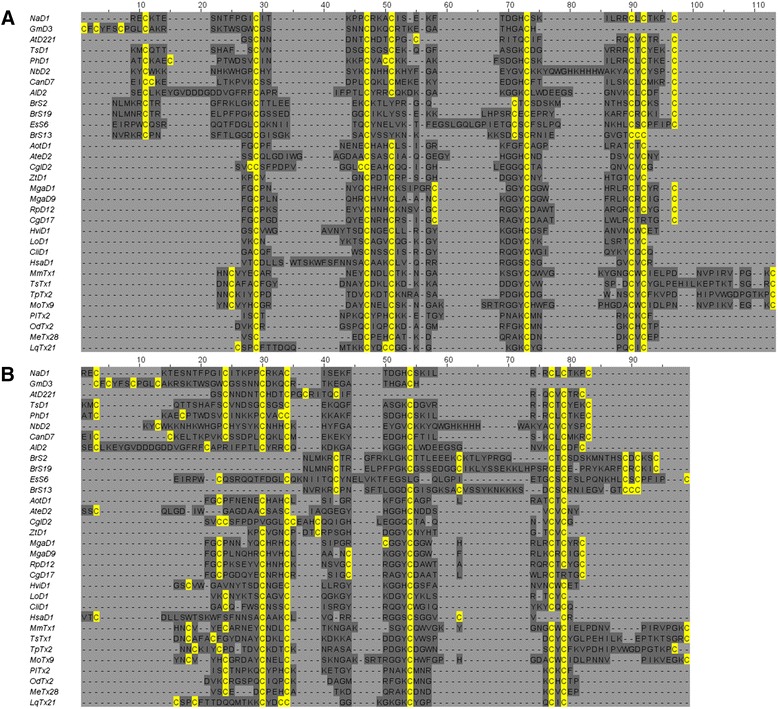
Alternative sequence alignment algorithms yield different results. It is therefore useful to quantify the similarities and differences between alternative alignments of the same sequences. These measurements can identify regions of consensus that are likely to be most informative in downstream analysis. They can also highlight systematic differences between alignments that relate to differences in the alignment algorithms themselves.
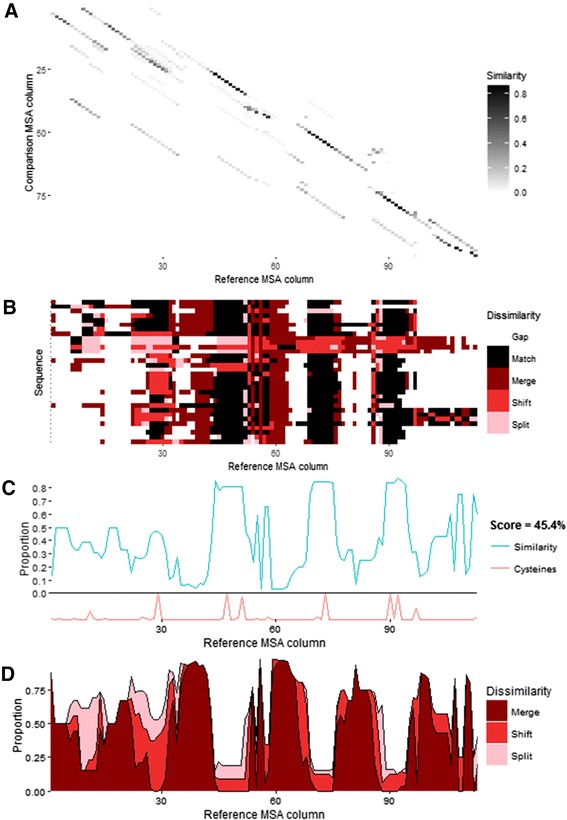
Here we present a simple method for aligning two alternative multiple sequence alignments to one another and assessing their similarity. Differences are categorised into merges, splits or shifts in one alignment relative to the other. A set of graphical visualisations allow for intuitive interpretation of the data.
AlignStat enables the easy one-off online use of MSA similarity comparisons or into R pipelines. The web-tool is available at AlignStat.Science.LaTrobe.edu.au. The R package, readme and example data are available on CRAN and GitHub.com/TS404/AlignStat.
不同的序列比对算法会产生不同的结果。因此,量化相同序列的不同比对之间的异同是很有用的。这些测量可以识别在下游分析中可能最具信息性的一致区域。它们还可以突出比对之间与比对算法本身差异相关的系统差异。
在这里,我们提出了一种简单的方法,用于将两个不同的多序列比对相互比对并评估它们的相似性。差异被分类为一个比对相对于另一个比对的合并、拆分或移位。一组图形可视化允许对数据进行直观解释。
AlignStat 使 MSA 相似性比较能够轻松地一次性在线使用或集成到 R 管道中。该网络工具可在 AlignStat.Science.LaTrobe.edu.au 上获取。R 包、自述文件和示例数据可在 CRAN 和 GitHub.com/TS404/AlignStat 上获取。