Amor David J, Marsh Ashley P L, Storey Elsdon, Tankard Rick, Gillies Greta, Delatycki Martin B, Pope Kate, Bromhead Catherine, Leventer Richard J, Bahlo Melanie, Lockhart Paul J
Murdoch Childrens Research Institute (D.J.A., A.P.L.M., G.G., M.B.D., K.P., R.J.L., P.J.L.), Royal Children's Hospital (D.J.A., M.B.D., R.J.L.), Parkville; Department of Paediatrics (D.J.A., A.P.L.M., M.B.D., C.B., R.J.L., P.J.L.), Department of Medical Biology (R.T., M.B.), The University of Melbourne; Department of Medicine (Neuroscience) (E.S.), Central Clinical School, Monash University; and Population Health and Immunity Division (R.T., M.B.), The Walter and Eliza Hall Institute of Medical Research, 1G Royal Parade, Parkville, Victoria, Australia.
Neurol Genet. 2016 Oct 18;2(6):e114. doi: 10.1212/NXG.0000000000000114. eCollection 2016 Dec.
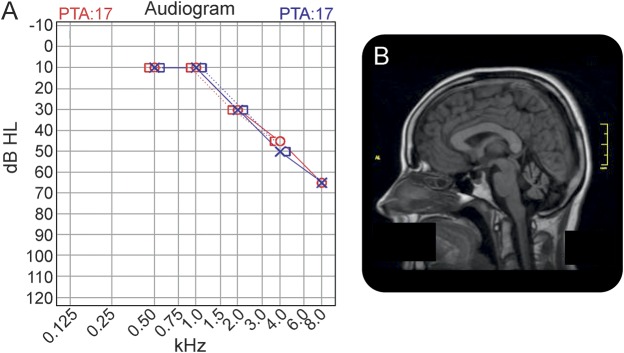
To determine the genetic cause of slowly progressive cerebellar ataxia, sensorineural deafness, and hypergonadotropic hypogonadism in 5 patients from 3 different families.
The patients comprised 2 sib pairs and 1 sporadic patient. Clinical assessment included history, physical examination, and brain MRI. Linkage analysis was performed separately on the 2 sets of sib pairs using single nucleotide polymorphism microarrays, followed by analysis of the intersection of the regions. Exome sequencing was performed on 1 affected patient with variant filtering and prioritization undertaken using these intersected regions.
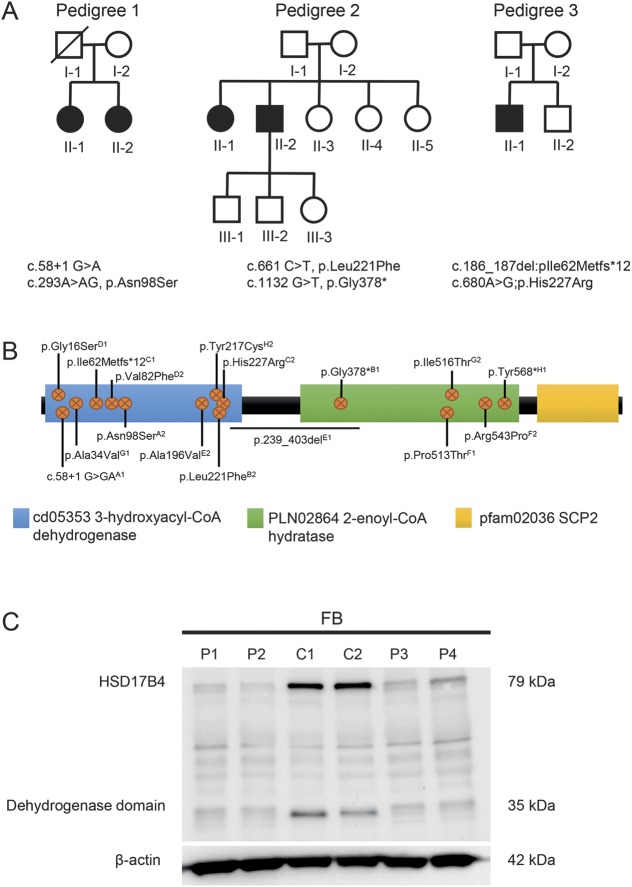
Using a combination of sequencing technologies, we identified compound heterozygous mutations in in all 5 affected patients. In all 3 families, peroxisomal D-bifunctional protein (DBP) deficiency was caused by compound heterozygosity for 1 nonsense/deletion mutation and 1 missense mutation.
We describe 5 patients with juvenile DBP deficiency from 3 different families, bringing the total number of reported patients to 14, from 8 families. This report broadens and consolidates the phenotype associated with juvenile DBP deficiency.
确定来自3个不同家庭的5例患者缓慢进展性小脑共济失调、感音神经性耳聋和高促性腺激素性性腺功能减退的遗传病因。
患者包括2对同胞对和1例散发性患者。临床评估包括病史、体格检查和脑部磁共振成像。使用单核苷酸多态性微阵列对2组同胞对分别进行连锁分析,随后分析这些区域的交集。对1例受累患者进行外显子组测序,并使用这些相交区域进行变异筛选和优先级排序。
通过结合多种测序技术,我们在所有5例受累患者中均鉴定出复合杂合突变。在所有3个家庭中,过氧化物酶体D-双功能蛋白(DBP)缺乏症是由1个无义/缺失突变和1个错义突变的复合杂合性引起的。
我们描述了来自3个不同家庭的5例青少年DBP缺乏症患者,使报告的患者总数达到来自8个家庭的14例。本报告拓宽并巩固了与青少年DBP缺乏症相关的表型。