Rosati Rayna, Patki Mugdha, Chari Venkatesh, Dakshnamurthy Selvakumar, McFall Thomas, Saxton Janice, Kidder Benjamin L, Shaw Peter E, Ratnam Manohar
From the Barbara Ann Karmanos Cancer Institute and Department of Oncology.
Wayne State University School of Medicine, Detroit, Michigan 48201-2013 and.
J Biol Chem. 2016 Dec 9;291(50):25983-25998. doi: 10.1074/jbc.M116.745596. Epub 2016 Oct 28.
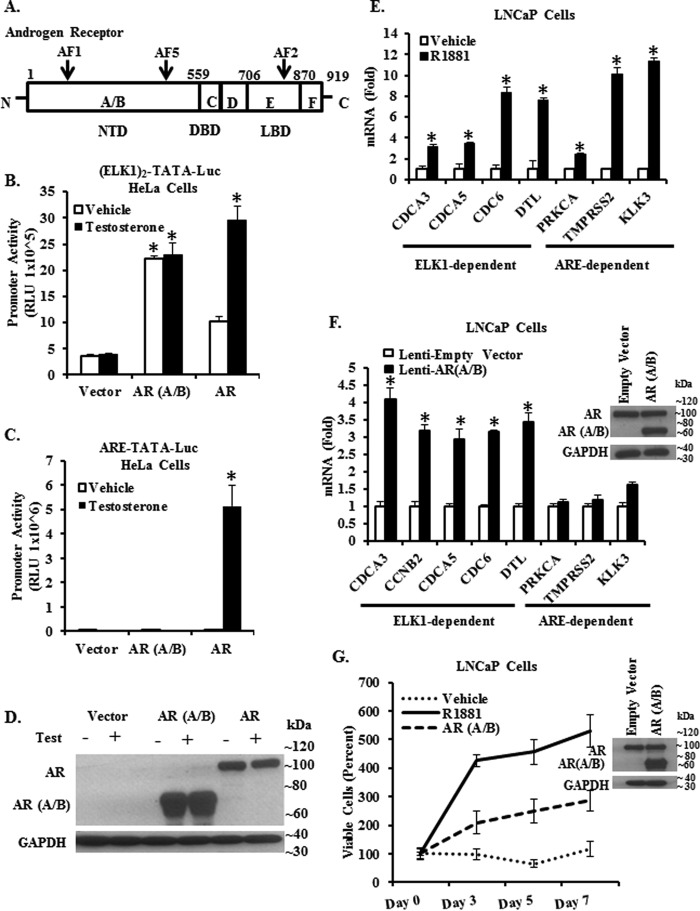
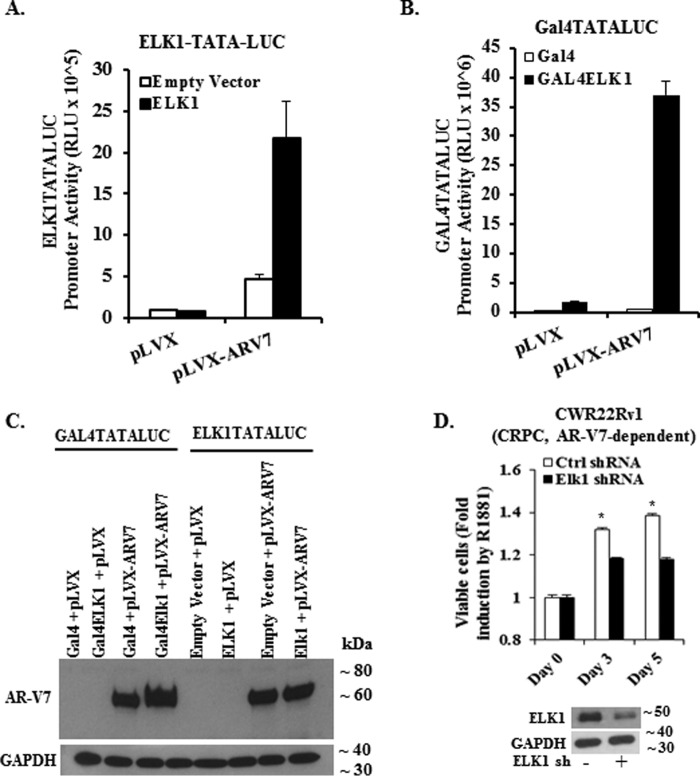
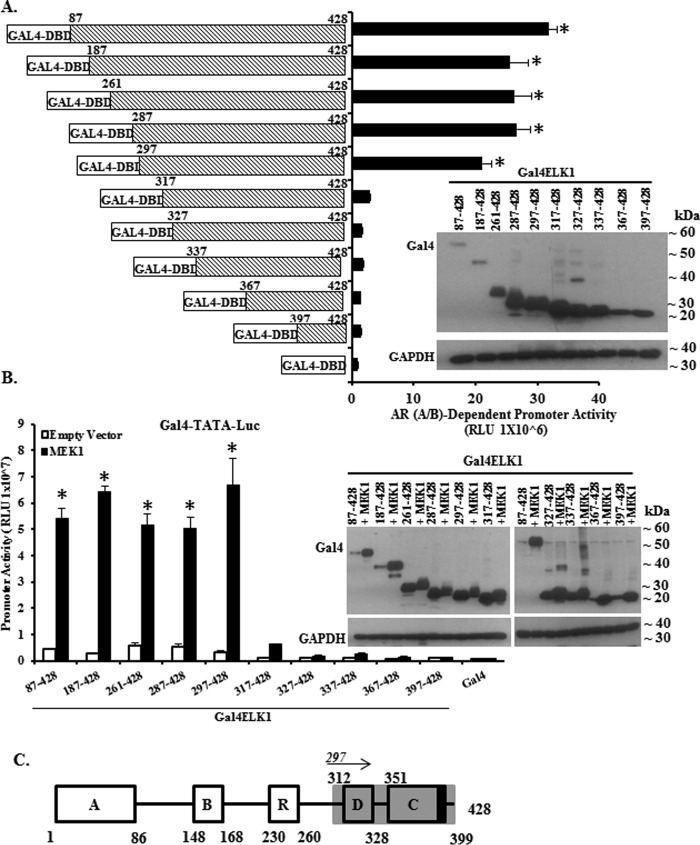
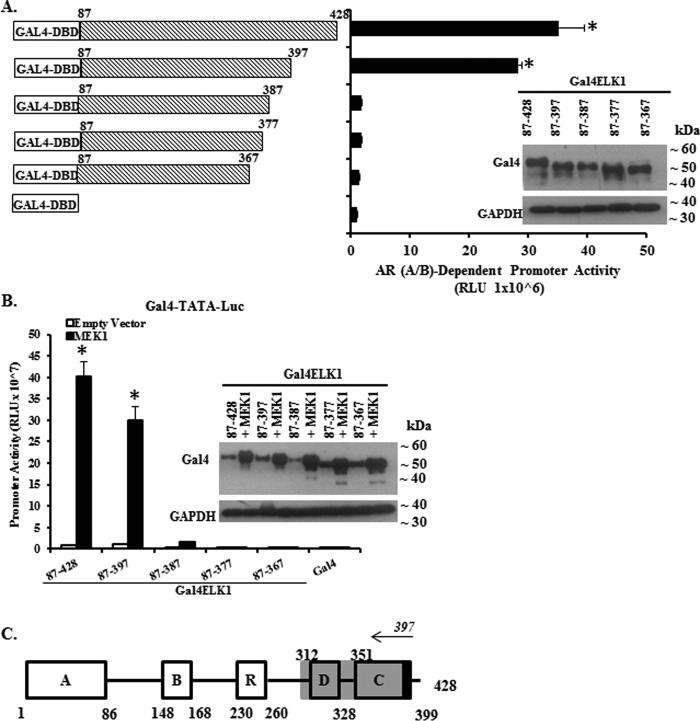
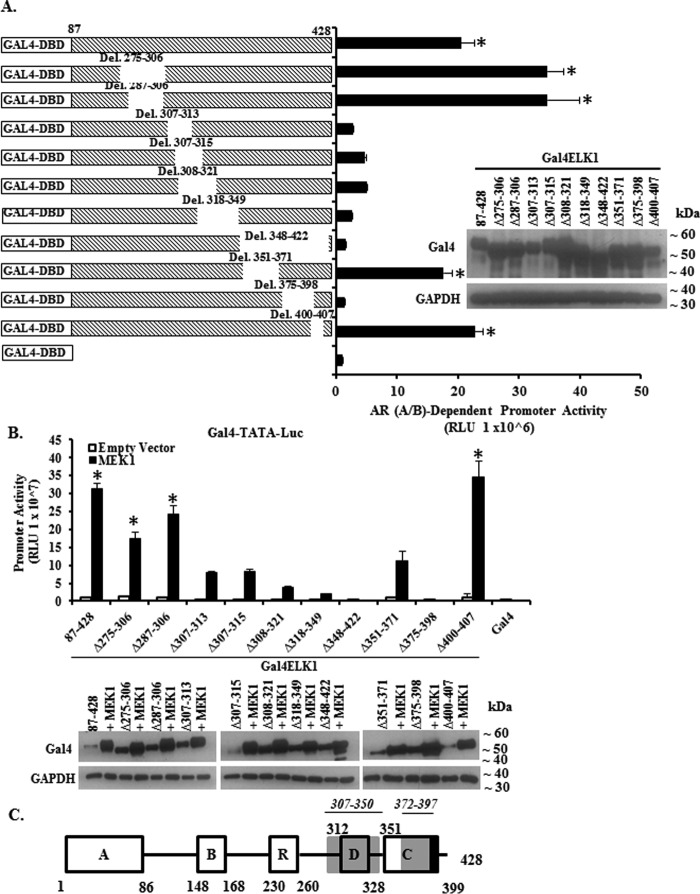
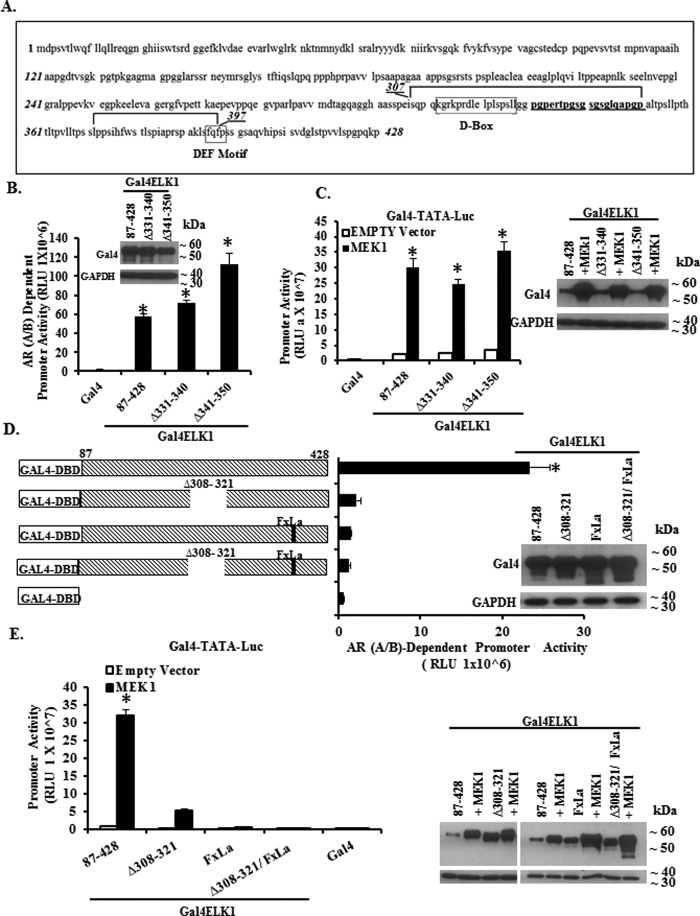
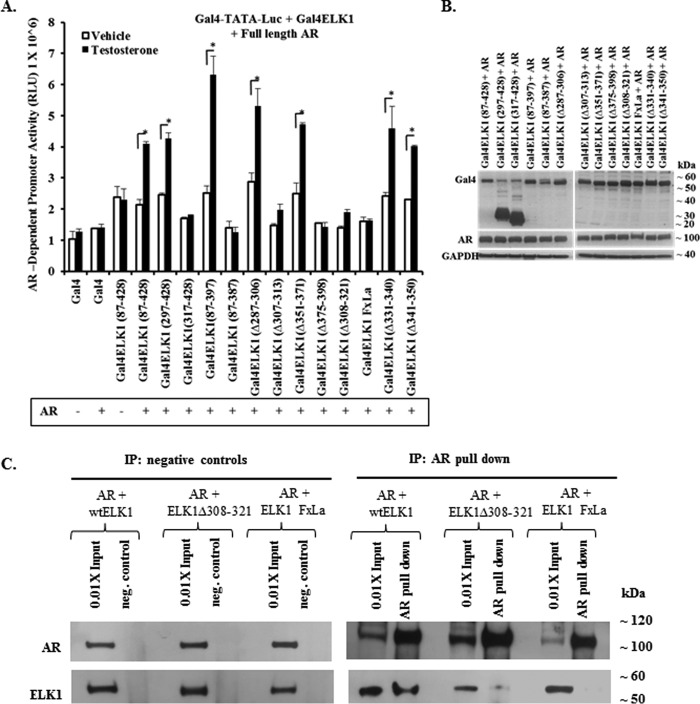
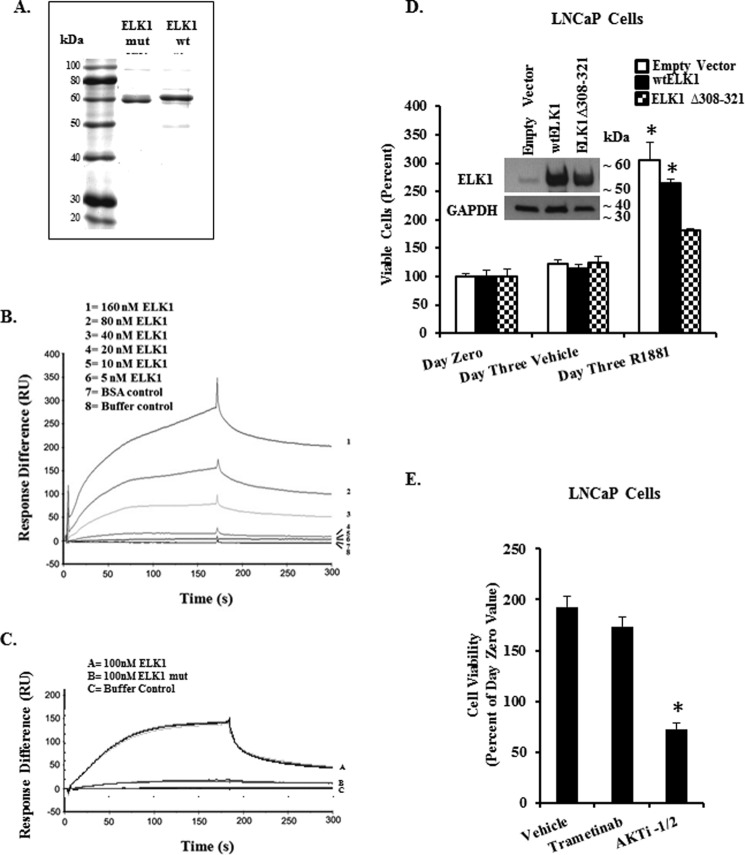
The ETS domain transcription factor ELK1 is in a repressive association with growth genes and is transiently activated through phosphorylation by ERK1/2. In prostate cancer (PCa) cells the androgen receptor (AR) is recruited by ELK1, via its amino-terminal domain (A/B), as a transcriptional co-activator, without ELK1 hyper-phosphorylation. Here we elucidate the structural basis of the interaction of AR with ELK1. The ELK1 polypeptide motifs required for co-activation by AR versus those required for activation of ELK1 by ERK were systematically mapped using a mammalian two-hybrid system and confirmed using a co-immunoprecipitation assay. The mapping precisely identified the two ERK-docking sites in ELK1, the D-box and the DEF (docking site for ERK, FXFP) motif, as the essential motifs for its cooperation with AR(A/B) or WTAR. In contrast, the transactivation domain in ELK1 was only required for activation by ERK. ELK1-mediated transcriptional activity of AR(A/B) was optimal in the absence of ELK1 binding partners, ERK1/2 and serum-response factor. Purified ELK1 and AR bound with a dissociation constant of 1.9 × 10 m A purified mutant ELK1 in which the D-box and DEF motifs were disrupted did not bind AR. An ELK1 mutant with deletion of the D-box region had a dominant-negative effect on androgen-dependent growth of PCa cells that were insensitive to MEK inhibition. This novel mechanism in which a nuclear receptor impinges on a signaling pathway by co-opting protein kinase docking sites to constitutively activate growth genes could enable rational design of a new class of targeted drug interventions.
ETS结构域转录因子ELK1与生长基因处于抑制性关联状态,并通过ERK1/2磷酸化而短暂激活。在前列腺癌细胞(PCa)中,雄激素受体(AR)通过其氨基末端结构域(A/B)被ELK1招募,作为转录共激活因子,而无需ELK1过度磷酸化。在此,我们阐明了AR与ELK1相互作用的结构基础。使用哺乳动物双杂交系统系统地绘制了AR共激活所需的ELK1多肽基序与ERK激活ELK1所需的基序,并通过免疫共沉淀试验进行了确认。该绘制精确地确定了ELK1中的两个ERK对接位点,即D框和DEF(ERK对接位点,FXFP)基序,是其与AR(A/B)或野生型AR合作的必需基序。相比之下,ELK1中的反式激活结构域仅在ERK激活时才需要。在没有ELK1结合伙伴、ERK1/2和血清反应因子的情况下,ELK1介导的AR(A/B)转录活性最佳。纯化的ELK1和AR以1.9×10 m的解离常数结合。纯化的D框和DEF基序被破坏的突变型ELK1不与AR结合。缺失D框区域的ELK1突变体对MEK抑制不敏感的PCa细胞的雄激素依赖性生长具有显性负效应。这种新机制,即核受体通过利用蛋白激酶对接位点来组成性激活生长基因从而影响信号通路,可能有助于合理设计一类新型靶向药物干预措施。