Jackson Kathryn L, Mbagwu Michael, Pacheco Jennifer A, Baldridge Abigail S, Viox Daniel J, Linneman James G, Shukla Sanjay K, Peissig Peggy L, Borthwick Kenneth M, Carrell David A, Bielinski Suzette J, Kirby Jacqueline C, Denny Joshua C, Mentch Frank D, Vazquez Lyam M, Rasmussen-Torvik Laura J, Kho Abel N
Feinberg School of Medicine, Northwestern University, Chicago, IL, USA.
Emory University School of Medicine, Atlanta, GA, USA.
BMC Infect Dis. 2016 Nov 17;16(1):684. doi: 10.1186/s12879-016-2020-2.
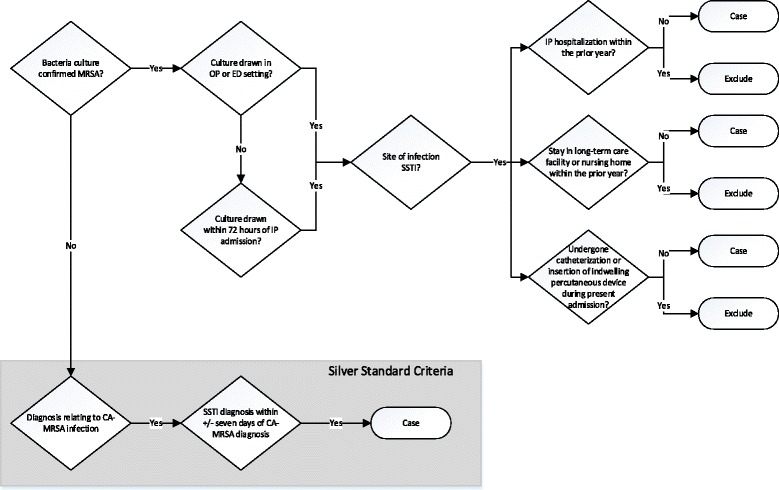
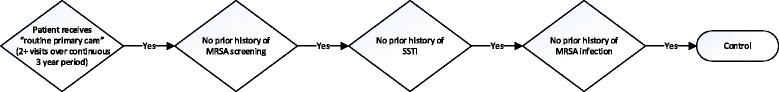
Community associated methicillin-resistant Staphylococcus aureus (CA-MRSA) is one of the most common causes of skin and soft tissue infections in the United States, and a variety of genetic host factors are suspected to be risk factors for recurrent infection. Based on the CDC definition, we have developed and validated an electronic health record (EHR) based CA-MRSA phenotype algorithm utilizing both structured and unstructured data.
The algorithm was validated at three eMERGE consortium sites, and positive predictive value, negative predictive value and sensitivity, were calculated. The algorithm was then run and data collected across seven total sites. The resulting data was used in GWAS analysis.
Across seven sites, the CA-MRSA phenotype algorithm identified a total of 349 cases and 7761 controls among the genotyped European and African American biobank populations. PPV ranged from 68 to 100% for cases and 96 to 100% for controls; sensitivity ranged from 94 to 100% for cases and 75 to 100% for controls. Frequency of cases in the populations varied widely by site. There were no plausible GWAS-significant (p < 5 E -8) findings.
Differences in EHR data representation and screening patterns across sites may have affected identification of cases and controls and accounted for varying frequencies across sites. Future work identifying these patterns is necessary.
社区获得性耐甲氧西林金黄色葡萄球菌(CA-MRSA)是美国皮肤和软组织感染最常见的病因之一,多种遗传宿主因素被怀疑是反复感染的危险因素。基于美国疾病控制与预防中心(CDC)的定义,我们开发并验证了一种利用结构化和非结构化数据的基于电子健康记录(EHR)的CA-MRSA表型算法。
该算法在三个电子医疗记录与基因组学(eMERGE)联盟站点进行了验证,并计算了阳性预测值、阴性预测值和敏感性。然后在总共七个站点运行该算法并收集数据。所得数据用于全基因组关联研究(GWAS)分析。
在七个站点中,CA-MRSA表型算法在基因分型的欧洲和非裔美国生物样本库人群中总共识别出349例病例和7761例对照。病例的阳性预测值范围为68%至100%,对照的阳性预测值范围为96%至100%;病例的敏感性范围为94%至100%,对照的敏感性范围为75%至100%。各站点人群中的病例频率差异很大。没有发现合理的全基因组关联研究显著(p < 5×10⁻⁸)结果。
各站点电子健康记录数据表示和筛查模式的差异可能影响了病例和对照的识别,并导致各站点频率不同。有必要开展进一步工作来识别这些模式。