Hsu Yung Chang, Coumar Mohane Selvaraj, Wang Wen-Chieh, Shiao Hui-Yi, Ke Yi-Yu, Lin Wen-Hsing, Kuo Ching-Chuan, Chang Chun-Wei, Kuo Fu-Ming, Chen Pei-Yi, Wang Sing-Yi, Li An-Siou, Chen Chun-Hwa, Kuo Po-Chu, Chen Ching-Ping, Wu Ming-Hsine, Huang Chen-Lung, Yen Kuei-Jung, Chang Yun-I, Hsu John T-A, Chen Chiung-Tong, Yeh Teng-Kuang, Song Jen-Shin, Shih Chuan, Hsieh Hsing-Pang
Institute of Biotechnology and Pharmaceutical Research, National Health Research Institutes, Zhunan, Taiwan, ROC.
Centre for Bioinformatics, School of Life Sciences, Pondicherry University, Kalapet, Puducherry, India.
Oncotarget. 2016 Dec 27;7(52):86239-86256. doi: 10.18632/oncotarget.13369.
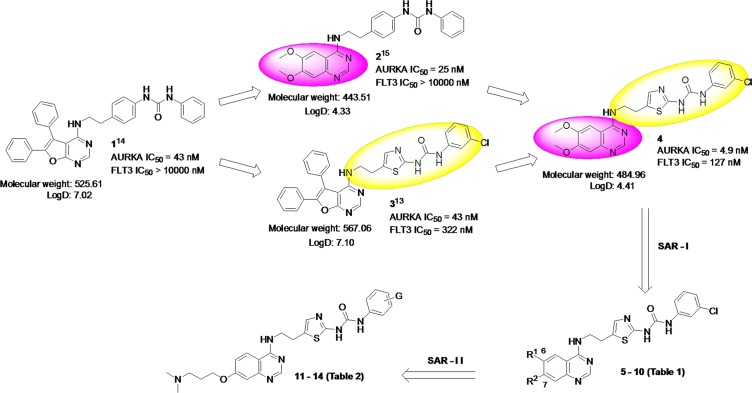
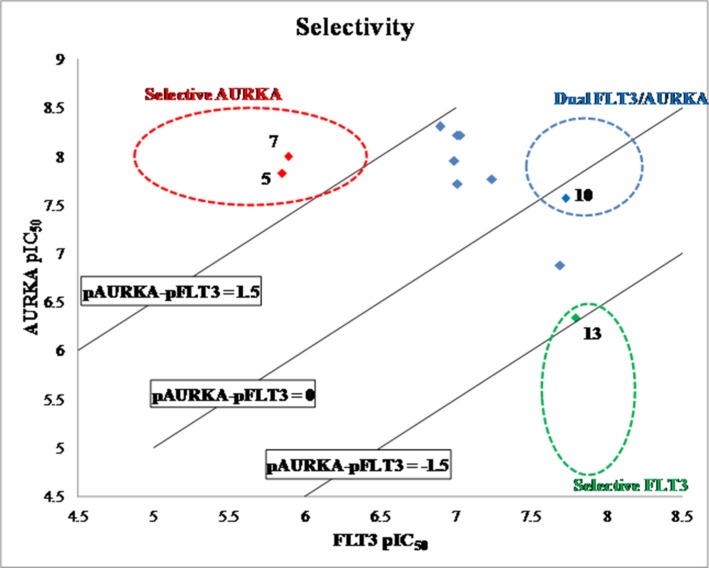
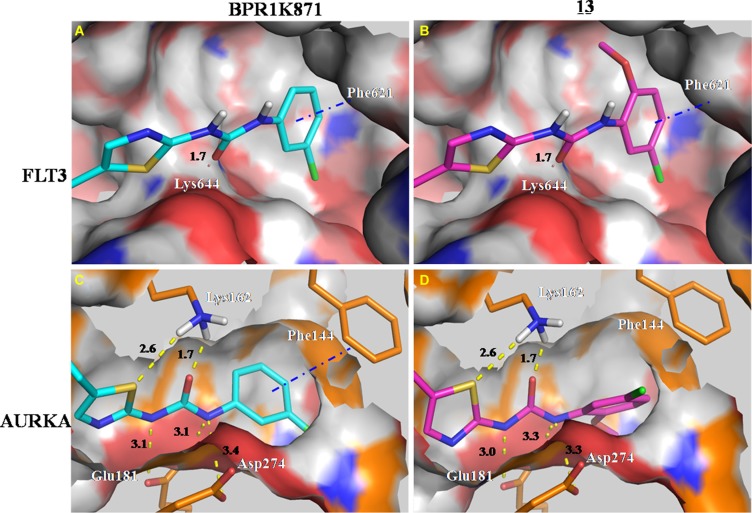
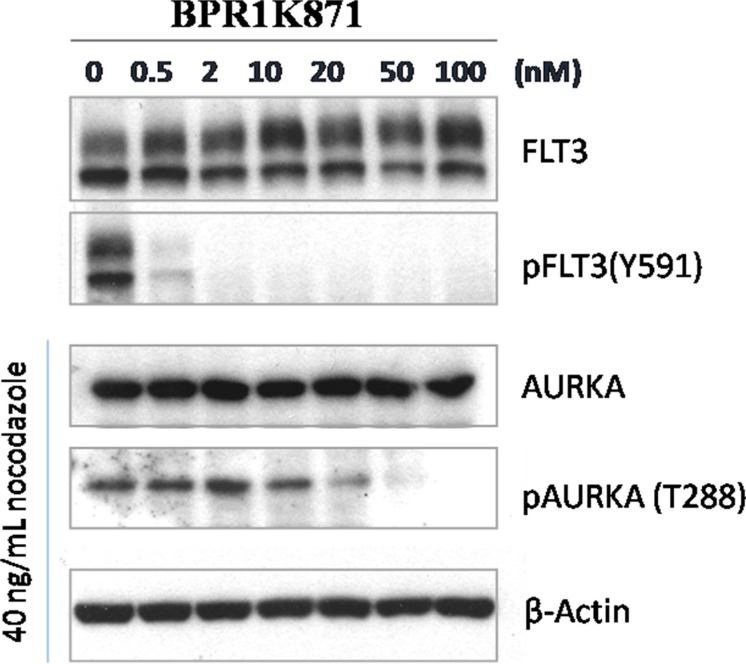
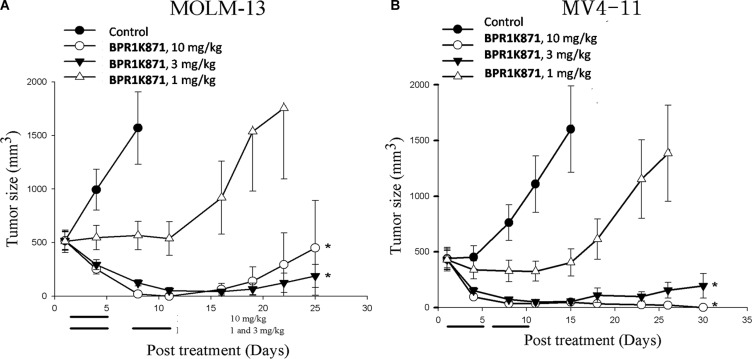
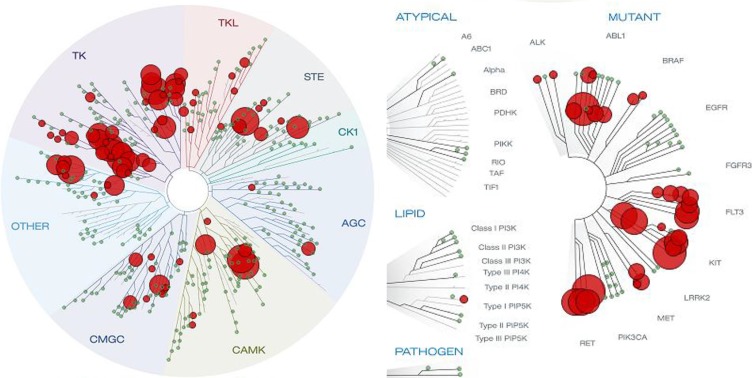
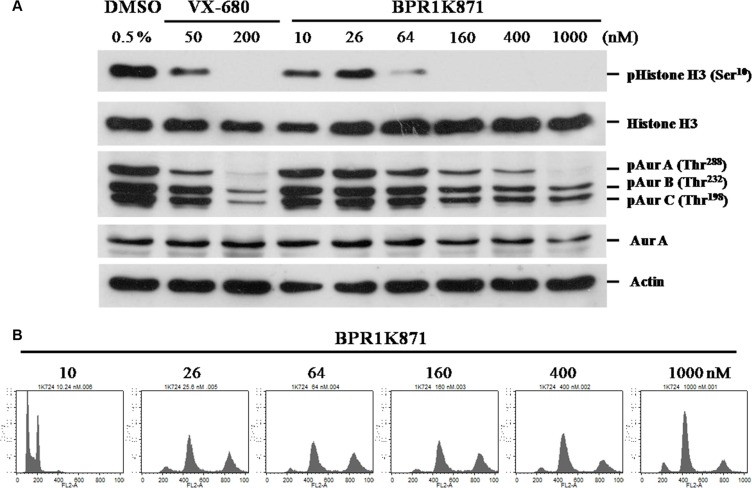
The design and synthesis of a quinazoline-based, multi-kinase inhibitor for the treatment of acute myeloid leukemia (AML) and other malignancies is reported. Based on the previously reported furanopyrimidine 3, quinazoline core containing lead 4 was synthesized and found to impart dual FLT3/AURKA inhibition (IC50 = 127/5 nM), as well as improved physicochemical properties. A detailed structure-activity relationship study of the lead 4 allowed FLT3 and AURKA inhibition to be finely tuned, resulting in AURKA selective (5 and 7; 100-fold selective over FLT3), FLT3 selective (13; 30-fold selective over AURKA) and dual FLT3/AURKA selective (BPR1K871; IC50 = 19/22 nM) agents. BPR1K871 showed potent anti-proliferative activities in MOLM-13 and MV4-11 AML cells (EC50 ~ 5 nM). Moreover, kinase profiling and cell-line profiling revealed BPR1K871 to be a potential multi-kinase inhibitor. Functional studies using western blot and DNA content analysis in MV4-11 and HCT-116 cell lines revealed FLT3 and AURKA/B target modulation inside the cells. In vivo efficacy in AML xenograft models (MOLM-13 and MV4-11), as well as in solid tumor models (COLO205 and Mia-PaCa2), led to the selection of BPR1K871 as a preclinical development candidate for anti-cancer therapy. Further detailed studies could help to investigate the full potential of BPR1K871 as a multi-kinase inhibitor.
报道了一种用于治疗急性髓性白血病(AML)和其他恶性肿瘤的喹唑啉类多激酶抑制剂的设计与合成。基于先前报道的呋喃嘧啶3,合成了含喹唑啉核心的先导化合物4,发现其具有双重FLT3/AURKA抑制作用(IC50 = 127/5 nM),以及改善的理化性质。对先导化合物4进行详细的构效关系研究,可对FLT3和AURKA抑制作用进行精细调节,从而得到AURKA选择性(化合物5和7;对FLT3的选择性为100倍)、FLT3选择性(化合物13;对AURKA的选择性为30倍)以及双重FLT3/AURKA选择性(BPR1K871;IC50 = 19/22 nM)的药物。BPR1K871在MOLM-13和MV4-11 AML细胞中表现出强大的抗增殖活性(EC50 ~ 5 nM)。此外,激酶谱分析和细胞系谱分析表明BPR1K871是一种潜在的多激酶抑制剂。在MV4-11和HCT-116细胞系中使用蛋白质印迹和DNA含量分析进行的功能研究揭示了细胞内FLT3和AURKA/B靶点的调节情况。在AML异种移植模型(MOLM-13和MV4-11)以及实体瘤模型(COLO205和Mia-PaCa2)中的体内疗效研究,使得BPR1K871被选为抗癌治疗的临床前开发候选药物。进一步的详细研究有助于探究BPR1K871作为多激酶抑制剂的全部潜力。