Duran Javier, Oyarce Cesar, Pavez Mario, Valladares Denisse, Basualto-Alarcon Carla, Lagos Daniel, Barrientos Genaro, Troncoso Mayarling Francisca, Ibarra Cristian, Estrada Manuel
Laboratorio de Endocrinología Celular, Programa de Fisiología y Biofísica, Instituto de Ciencias Biomédicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile.
Programa de Anatomía y Biología del Desarrollo, Instituto de Ciencias Biomédicas, Facultad de Medicina, Universidad de Chile, Santiago, Chile.
PLoS One. 2016 Dec 15;11(12):e0168255. doi: 10.1371/journal.pone.0168255. eCollection 2016.
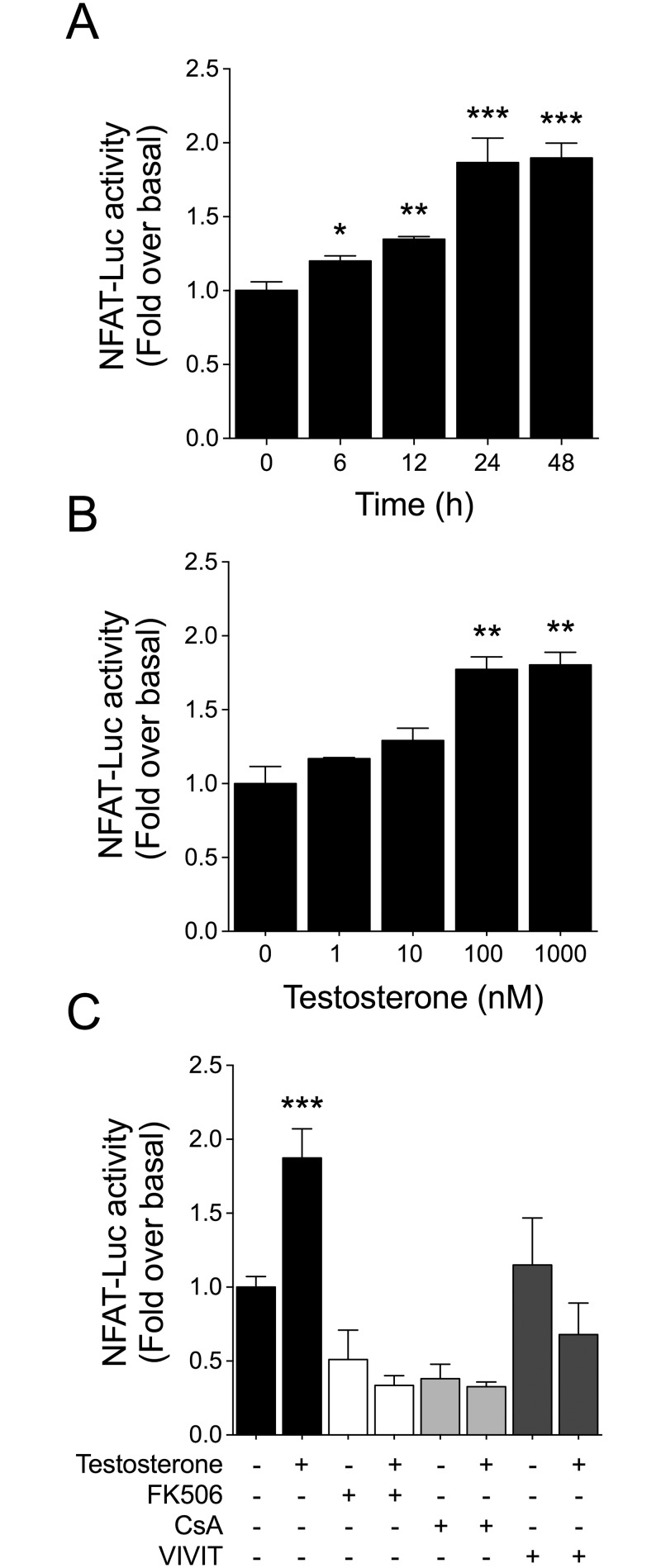
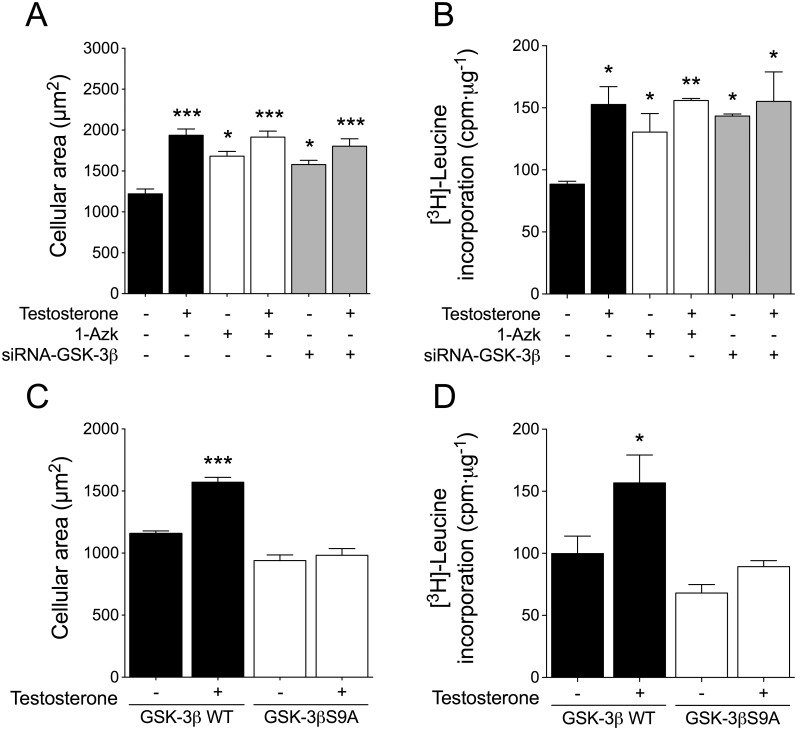
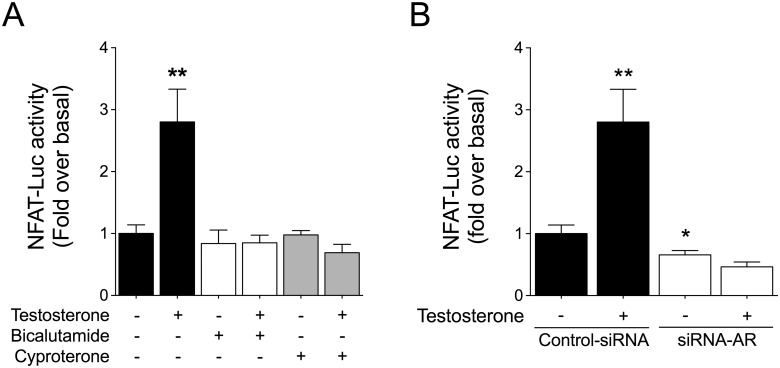
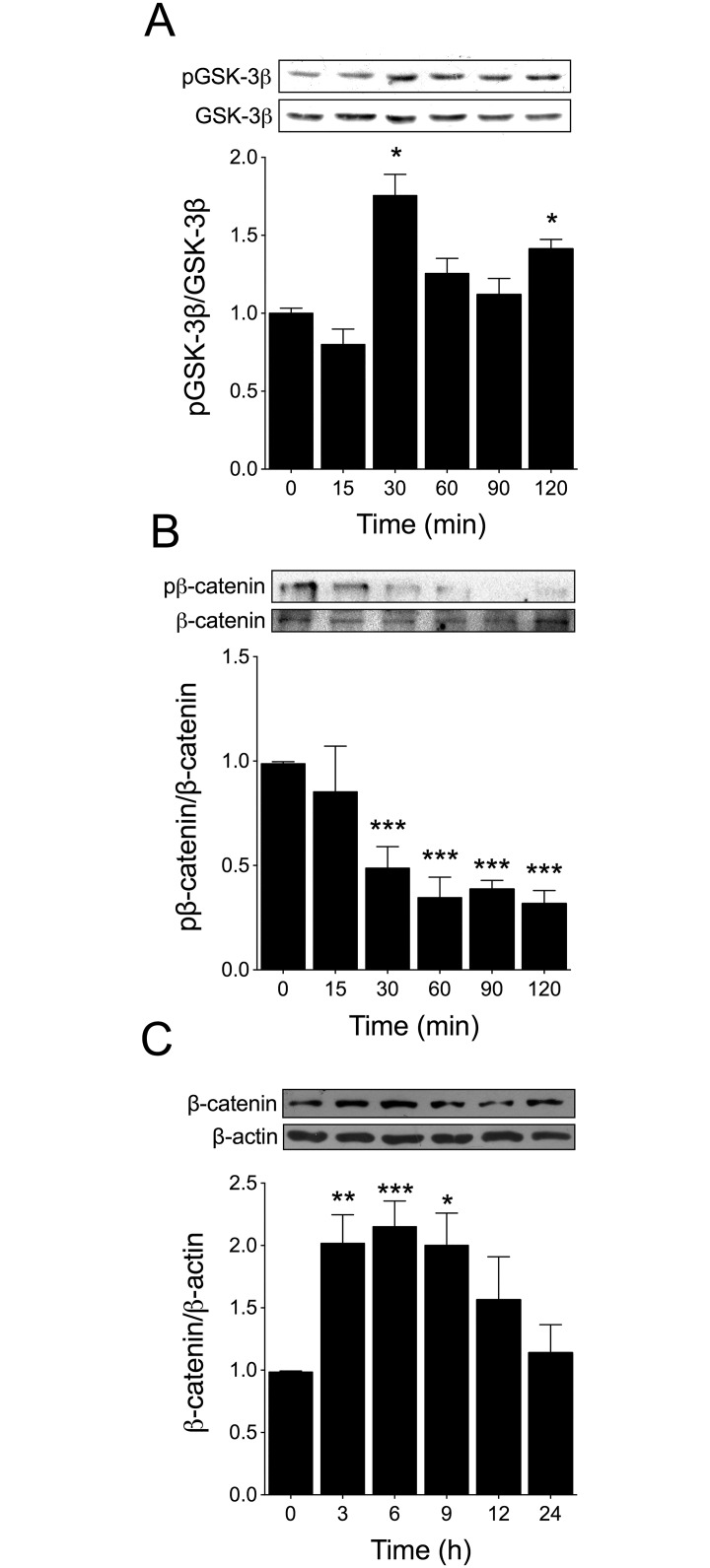
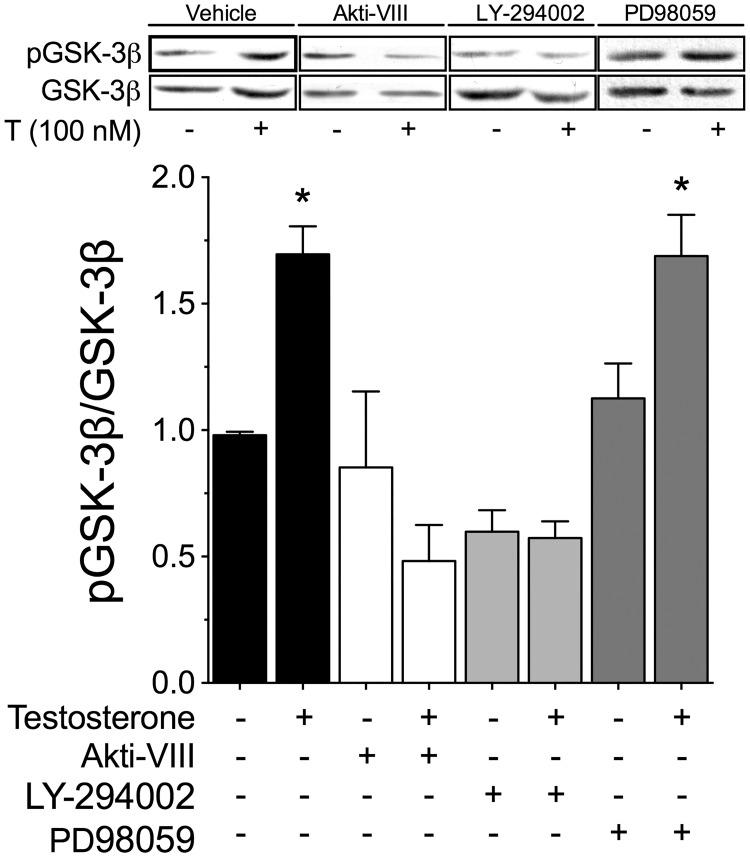
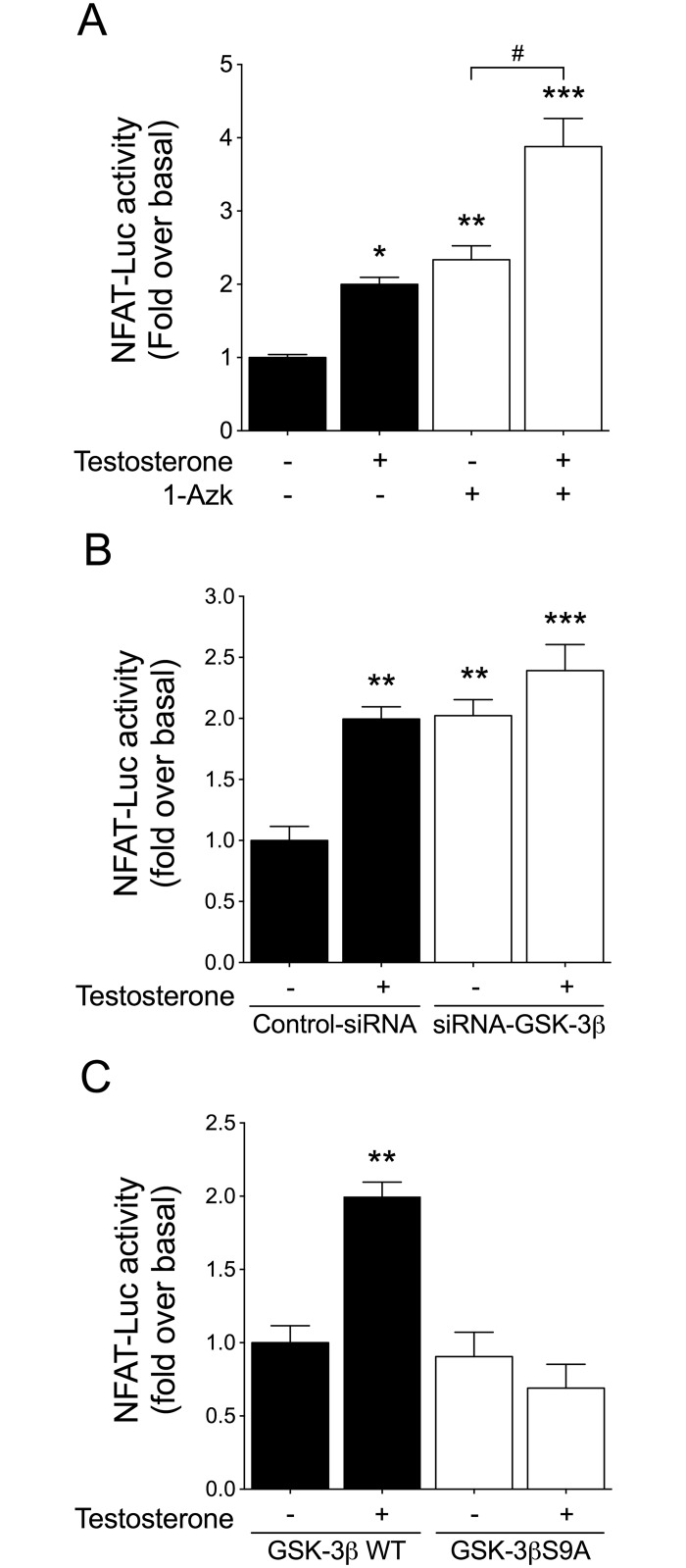
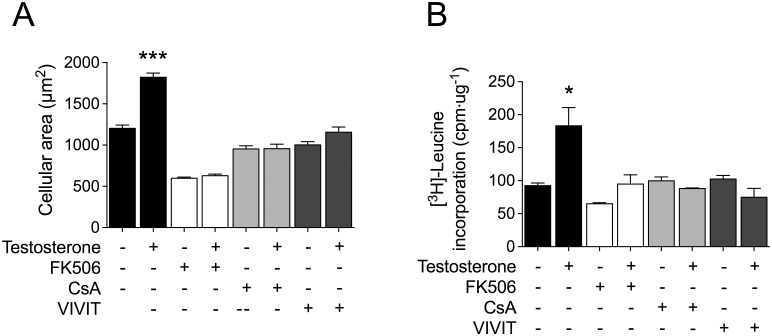
Testosterone induces cardiac hypertrophy through a mechanism that involves a concerted crosstalk between cytosolic and nuclear signaling pathways. Nuclear factor of activated T-cells (NFAT) is associated with the promotion of cardiac hypertrophy, glycogen synthase kinase-3β (GSK-3β) is considered to function as a negative regulator, mainly by modulating NFAT activity. However, the role played by calcineurin-NFAT and GSK-3β signaling in testosterone-induced cardiac hypertrophy has remained unknown. Here, we determined that testosterone stimulates cardiac myocyte hypertrophy through NFAT activation and GSK-3β inhibition. Testosterone increased the activity of NFAT-luciferase (NFAT-Luc) in a time- and dose-dependent manner, with the activity peaking after 24 h of stimulation with 100 nM testosterone. NFAT-Luc activity induced by testosterone was blocked by the calcineurin inhibitors FK506 and cyclosporine A and by 11R-VIVIT, a specific peptide inhibitor of NFAT. Conversely, testosterone inhibited GSK-3β activity as determined by increased GSK-3β phosphorylation at Ser9 and β-catenin protein accumulation, and also by reduction in β-catenin phosphorylation at residues Ser33, Ser37, and Thr41. GSK-3β inhibition with 1-azakenpaullone or a GSK-3β-targeting siRNA increased NFAT-Luc activity, whereas overexpression of a constitutively active GSK-3β mutant (GSK-3βS9A) inhibited NFAT-Luc activation mediated by testosterone. Testosterone-induced cardiac myocyte hypertrophy was established by increased cardiac myocyte size and [3H]-leucine incorporation (as a measurement of cellular protein synthesis). Calcineurin-NFAT inhibition abolished and GSK-3β inhibition promoted the hypertrophy stimulated by testosterone. GSK-3β activation by GSK-3βS9A blocked the increase of hypertrophic markers induced by testosterone. Moreover, inhibition of intracellular androgen receptor prevented testosterone-induced NFAT-Luc activation. Collectively, these results suggest that cardiac myocyte hypertrophy induced by testosterone involves a cooperative mechanism that links androgen signaling with the recruitment of NFAT through calcineurin activation and GSK-3β inhibition.
睾酮通过一种涉及胞质和核信号通路协同串扰的机制诱导心脏肥大。活化T细胞核因子(NFAT)与心脏肥大的促进相关,糖原合酶激酶-3β(GSK-3β)被认为主要通过调节NFAT活性而发挥负调节作用。然而,钙调神经磷酸酶-NFAT和GSK-3β信号在睾酮诱导的心脏肥大中所起的作用仍不清楚。在此,我们确定睾酮通过NFAT激活和GSK-3β抑制刺激心肌细胞肥大。睾酮以时间和剂量依赖性方式增加NFAT荧光素酶(NFAT-Luc)的活性,在用100 nM睾酮刺激24小时后活性达到峰值。睾酮诱导的NFAT-Luc活性被钙调神经磷酸酶抑制剂FK506和环孢素A以及NFAT的特异性肽抑制剂11R-VIVIT阻断。相反,如通过Ser9处GSK-3β磷酸化增加和β-连环蛋白蛋白积累所确定,以及通过Ser33、Ser37和Thr41残基处β-连环蛋白磷酸化减少所确定,睾酮抑制GSK-3β活性。用1-氮杂肯帕乌洛酮或靶向GSK-3β的小干扰RNA抑制GSK-3β增加NFAT-Luc活性,而组成型活性GSK-3β突变体(GSK-3βS9A)的过表达抑制睾酮介导的NFAT-Luc激活。通过增加心肌细胞大小和[3H]-亮氨酸掺入(作为细胞蛋白质合成的一种测量)确定了睾酮诱导的心肌细胞肥大。钙调神经磷酸酶-NFAT抑制消除了睾酮刺激的肥大,而GSK-3β抑制促进了这种肥大。GSK-3βS9A激活GSK-3β阻断了睾酮诱导的肥大标志物的增加。此外,抑制细胞内雄激素受体可防止睾酮诱导的NFAT-Luc激活。总体而言,这些结果表明,睾酮诱导的心肌细胞肥大涉及一种协同机制,该机制通过钙调神经磷酸酶激活和GSK-3β抑制将雄激素信号与NFAT募集联系起来。