Zhang Changsheng, Bell David, Harger Matthew, Ren Pengyu
Department of Biomedical Engineering, The University of Texas at Austin , Austin, Texas 78712, United States.
J Chem Theory Comput. 2017 Feb 14;13(2):666-678. doi: 10.1021/acs.jctc.6b00918. Epub 2017 Jan 13.
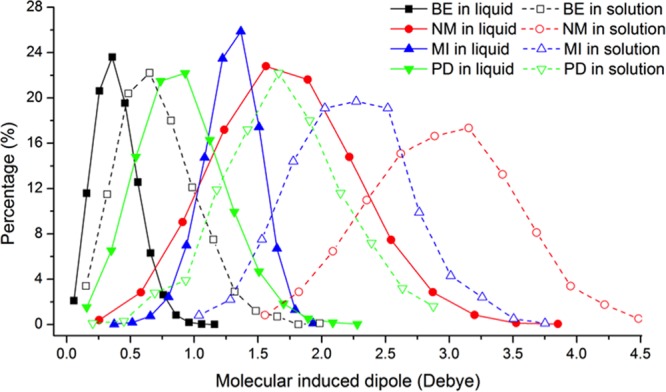
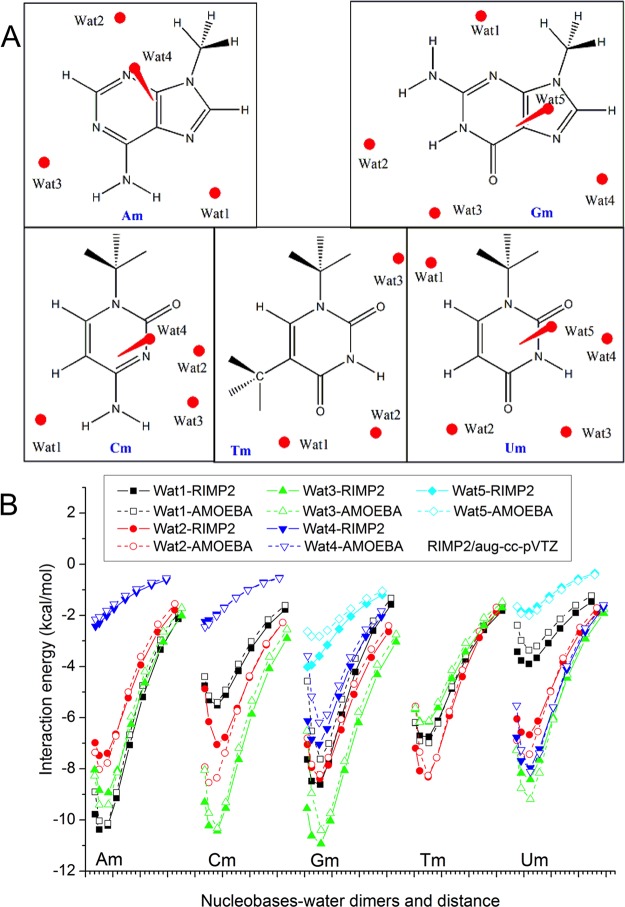
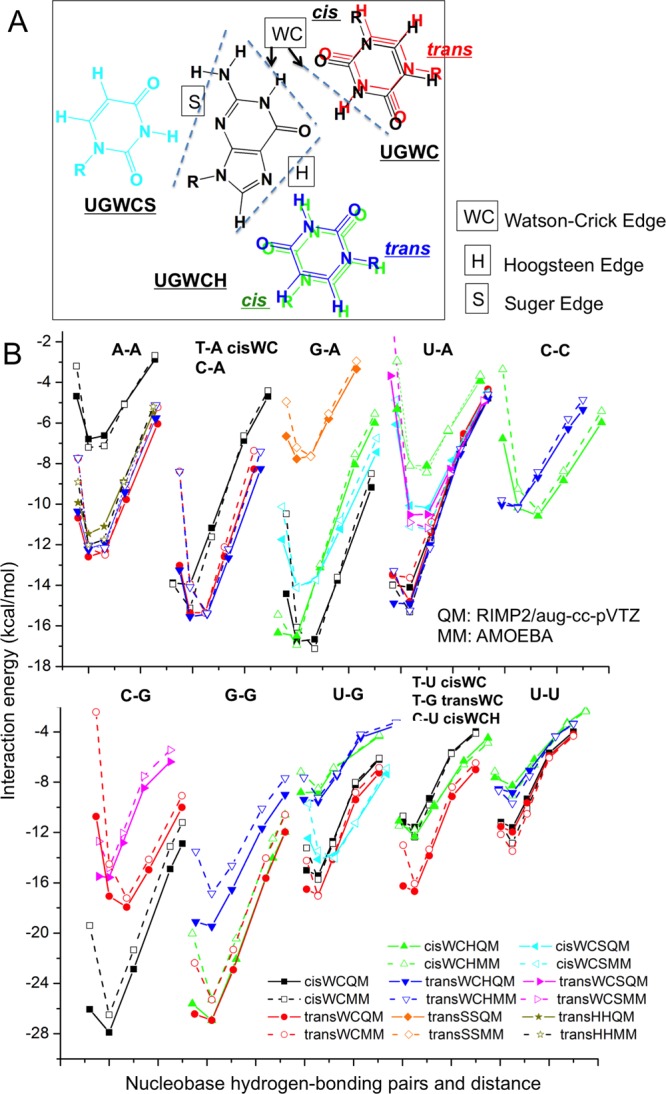
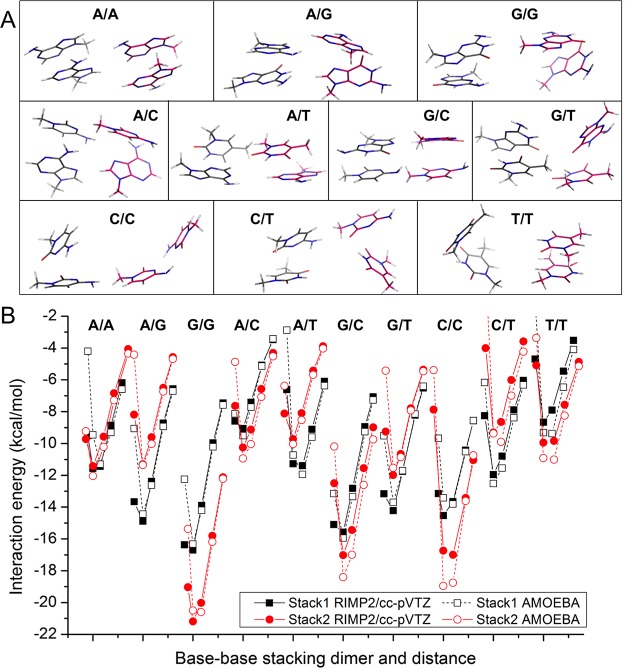
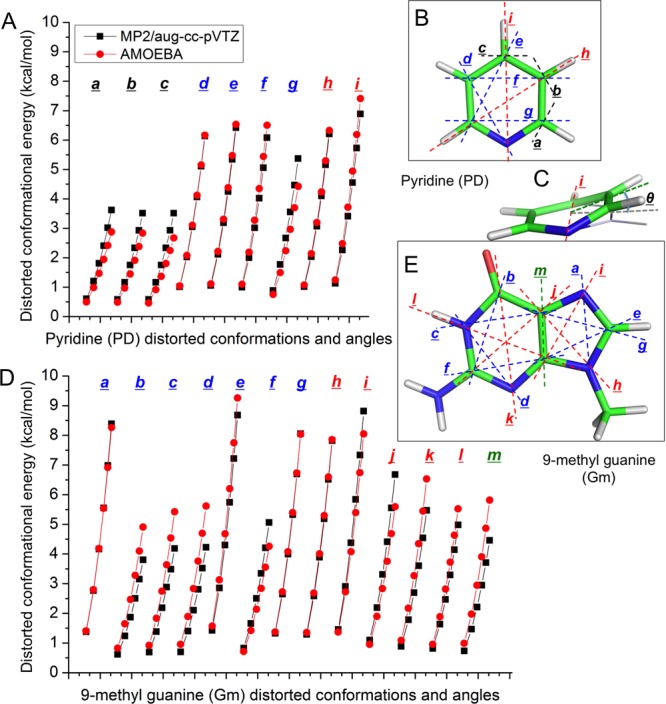
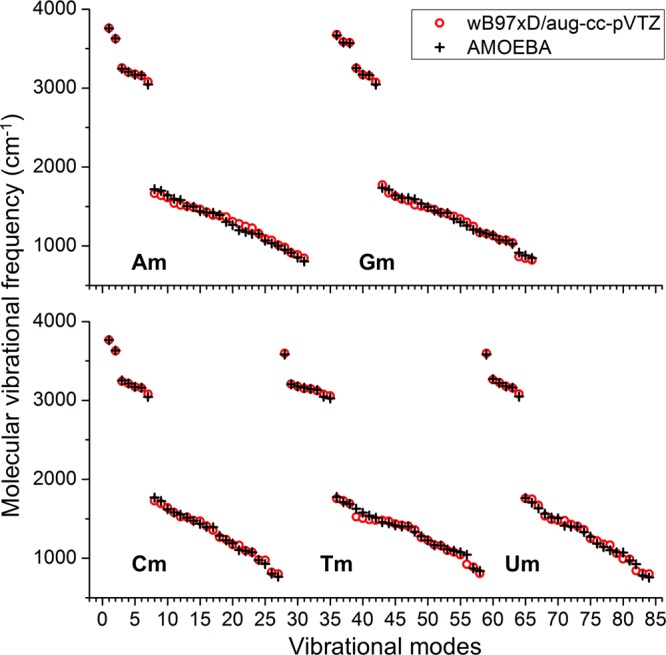
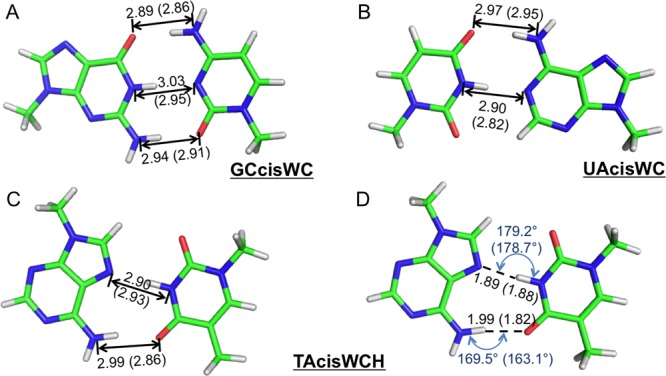
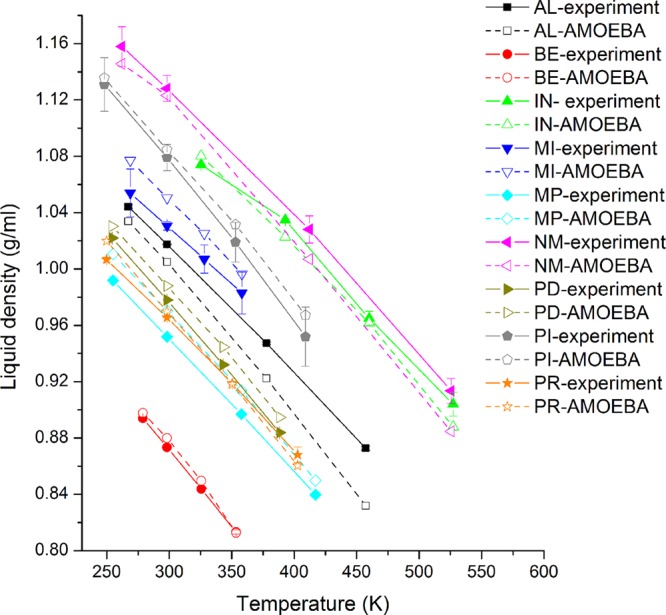
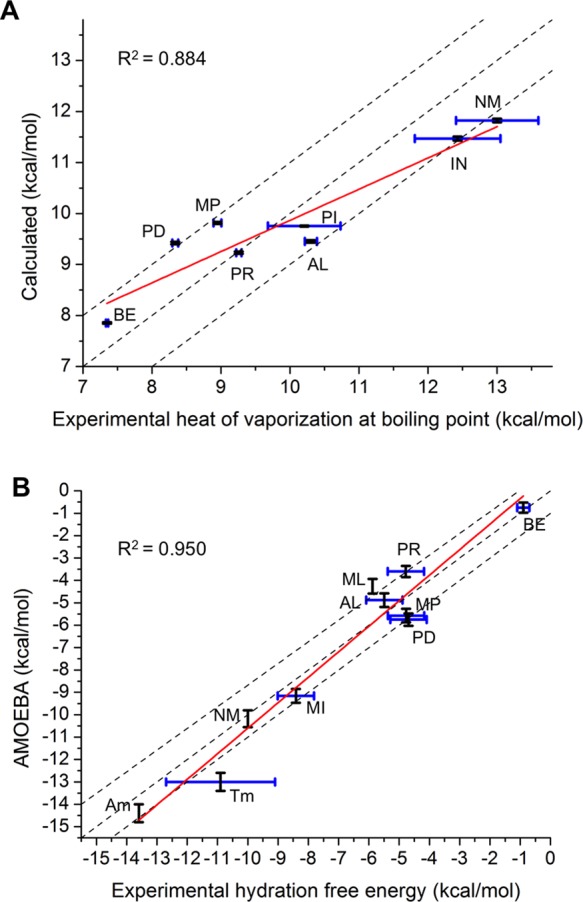
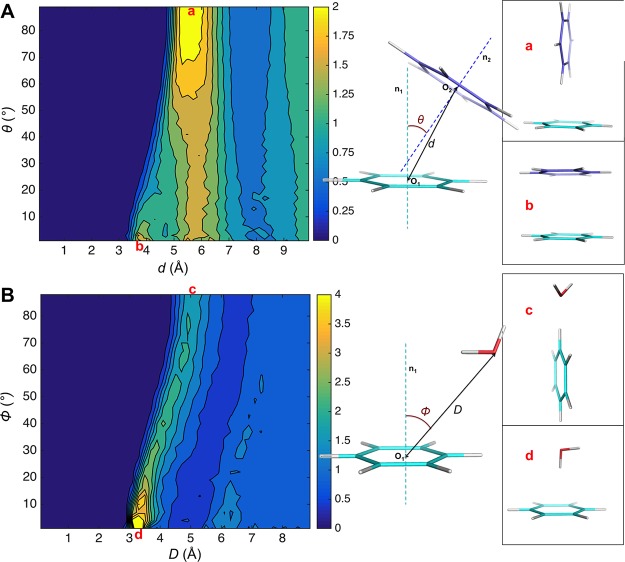
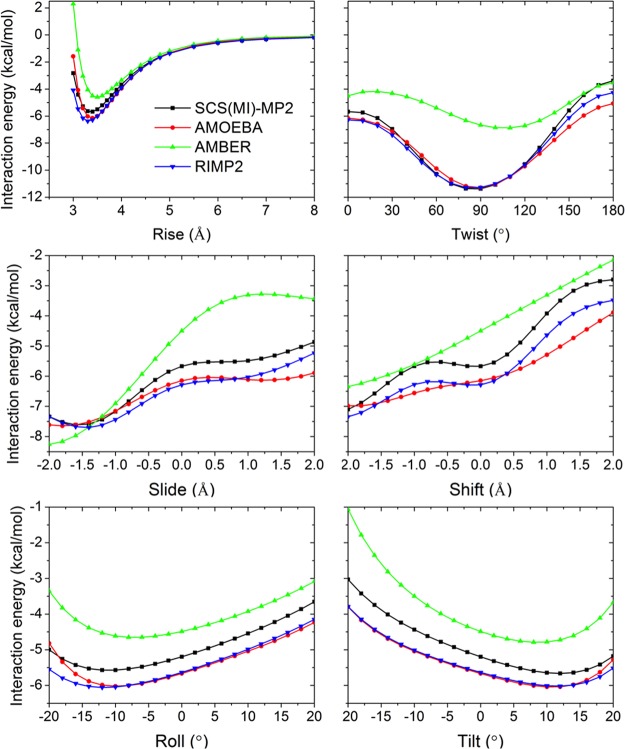
Aromatic molecules with π electrons are commonly involved in chemical and biological recognitions. For example, nucleobases play central roles in DNA/RNA structure and their interactions with proteins. The delocalization of the π electrons is responsible for the high polarizability of aromatic molecules. In this work, the AMOEBA force field has been developed and applied to 5 regular nucleobases and 12 aromatic molecules. The permanent electrostatic energy is expressed as atomic multipole interactions between atom pairs, and many-body polarization is accounted for by mutually induced atomic dipoles. We have systematically investigated aromatic ring stacking and aromatic-water interactions for nucleobases and aromatic molecules, as well as base-base hydrogen-bonding pair interactions, all at various distances and orientations. van der Waals parameters were determined by comparison to the quantum mechanical interaction energy of these dimers and fine-tuned using condensed phase simulation. By comparing to quantum mechanical calculations, we show that the resulting classical potential is able to accurately describe molecular polarizability, molecular vibrational frequency, and dimer interaction energy of these aromatic systems. Condensed phase properties, including hydration free energy, liquid density, and heat of vaporization, are also in good overall agreement with experimental values. The structures of benzene liquid phase and benzene-water solution were also investigated by simulation and compared with experimental and PDB structure derived statistical results.
具有π电子的芳香分子通常参与化学和生物识别过程。例如,核碱基在DNA/RNA结构及其与蛋白质的相互作用中起着核心作用。π电子的离域作用导致了芳香分子的高极化率。在这项工作中,开发了AMOEBA力场并将其应用于5种常规核碱基和12种芳香分子。永久静电能表示为原子对之间的原子多极相互作用,多体极化则由相互诱导的原子偶极来描述。我们系统地研究了核碱基和芳香分子的芳香环堆积和芳香-水相互作用,以及碱基-碱基氢键对相互作用,所有这些都是在不同的距离和取向下进行的。通过与这些二聚体的量子力学相互作用能进行比较来确定范德华参数,并使用凝聚相模拟进行微调。通过与量子力学计算结果进行比较,我们表明所得到的经典势能够准确描述这些芳香体系的分子极化率、分子振动频率和二聚体相互作用能。凝聚相性质,包括水合自由能、液体密度和汽化热,总体上也与实验值吻合良好。还通过模拟研究了苯液相和苯-水溶液的结构,并与实验结果以及从PDB结构得出的统计结果进行了比较。