Jeong Eun Il, Chung Hae Won, Lee Won Jea, Kim Seo-Hyun, Kim Hyunjoo, Choi Seon-Guk, Jung Yong-Keun
Global Research Laboratory, Department of Biological Science, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-747, Korea.
Cell Death Dis. 2016 Dec 29;7(12):e2573. doi: 10.1038/cddis.2016.428.
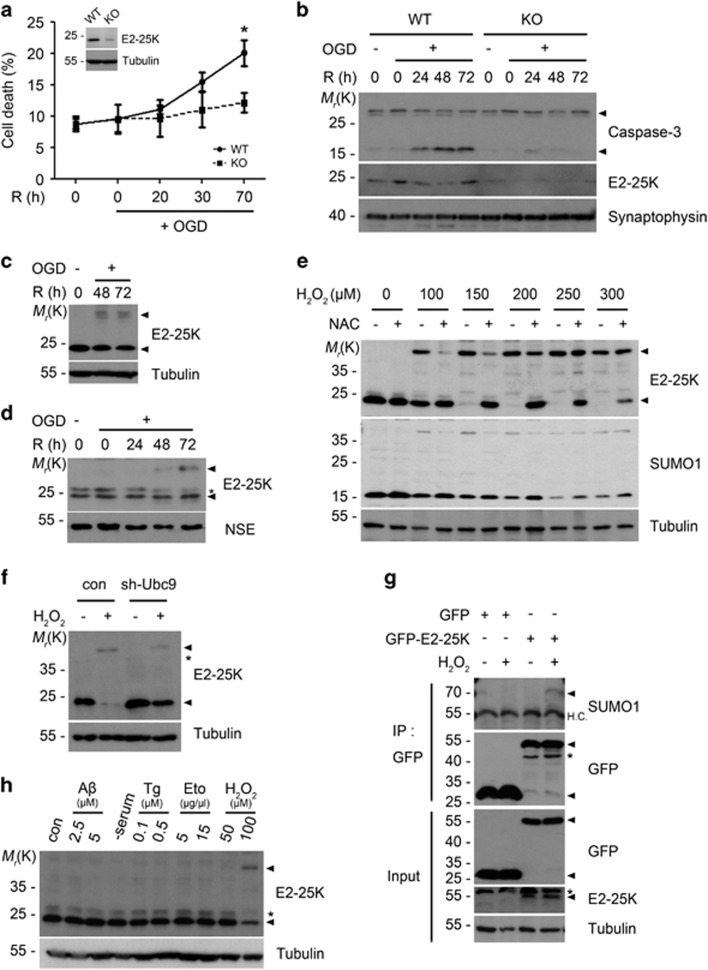
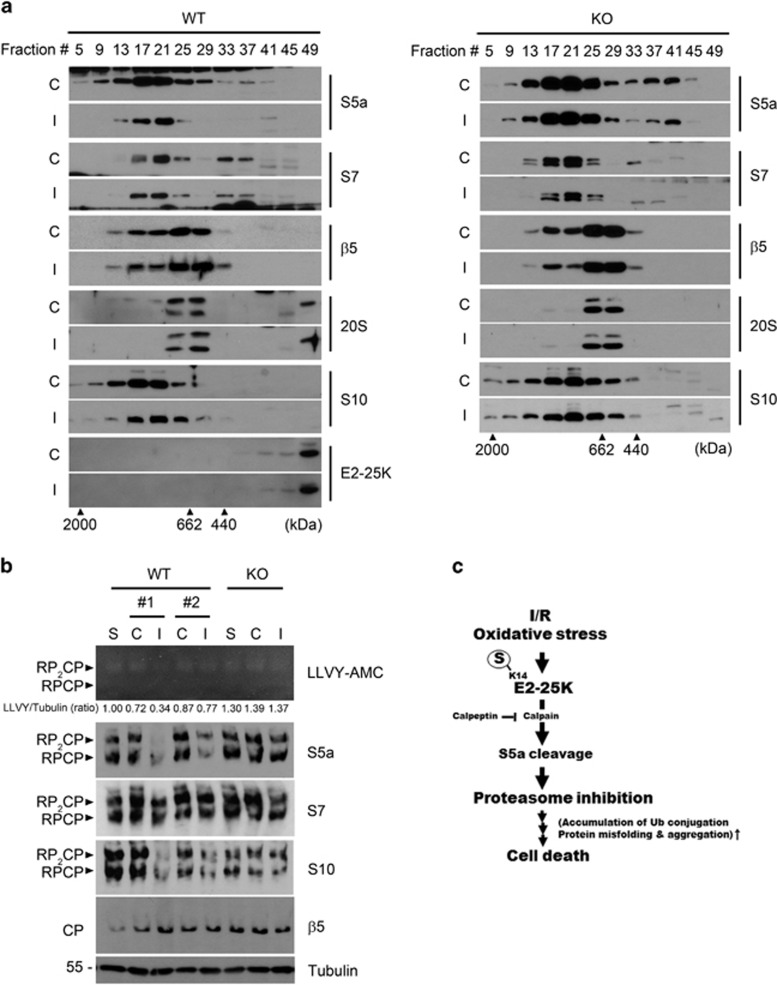
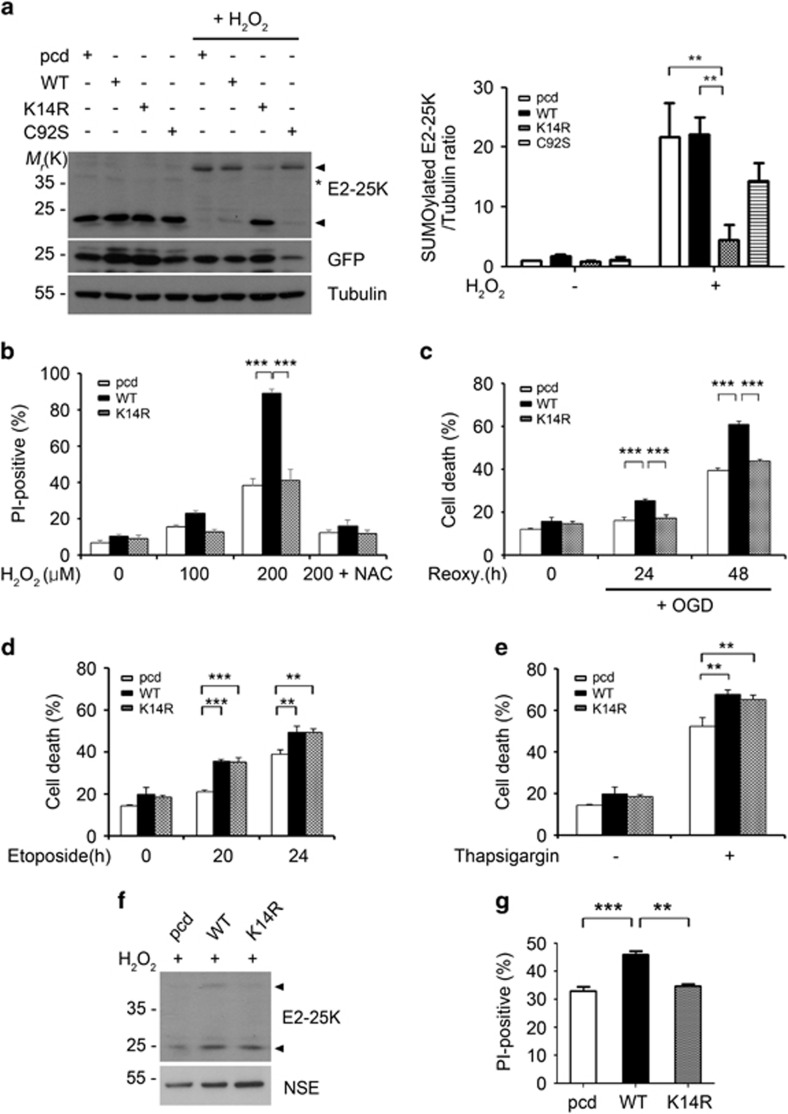
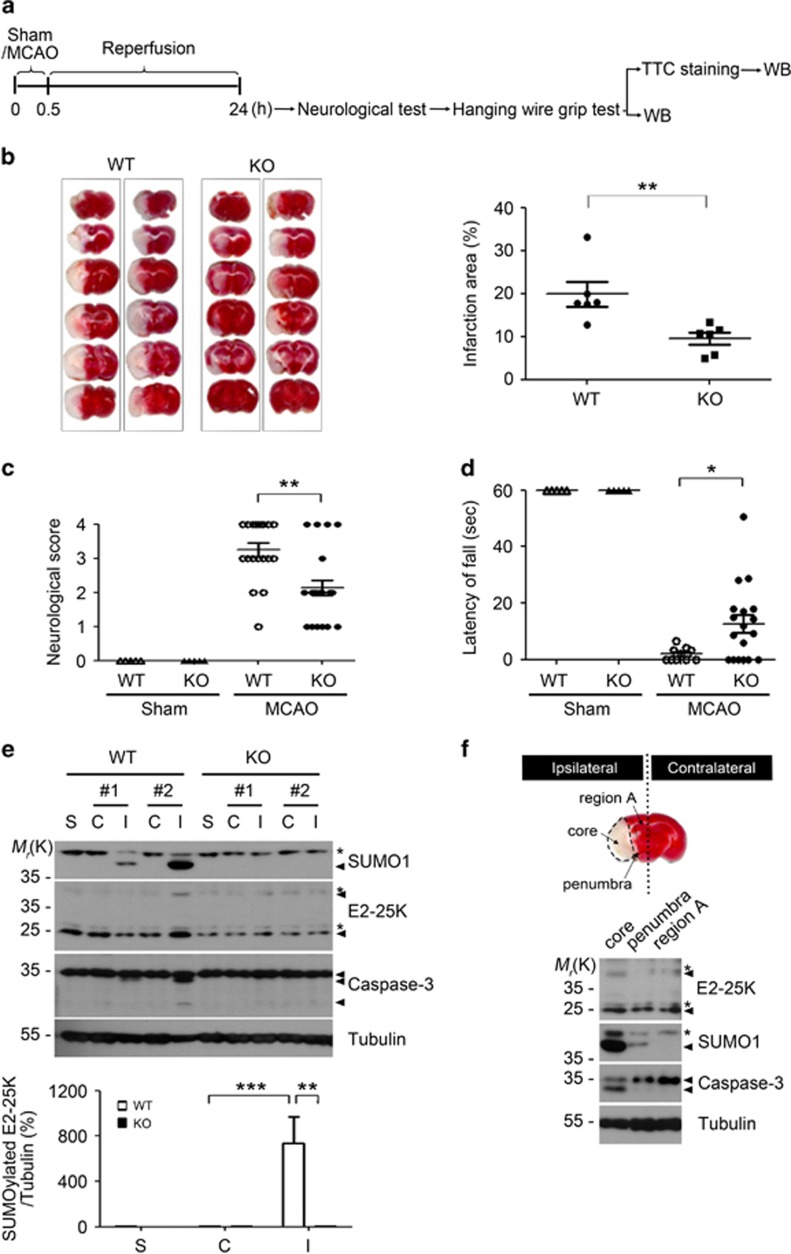
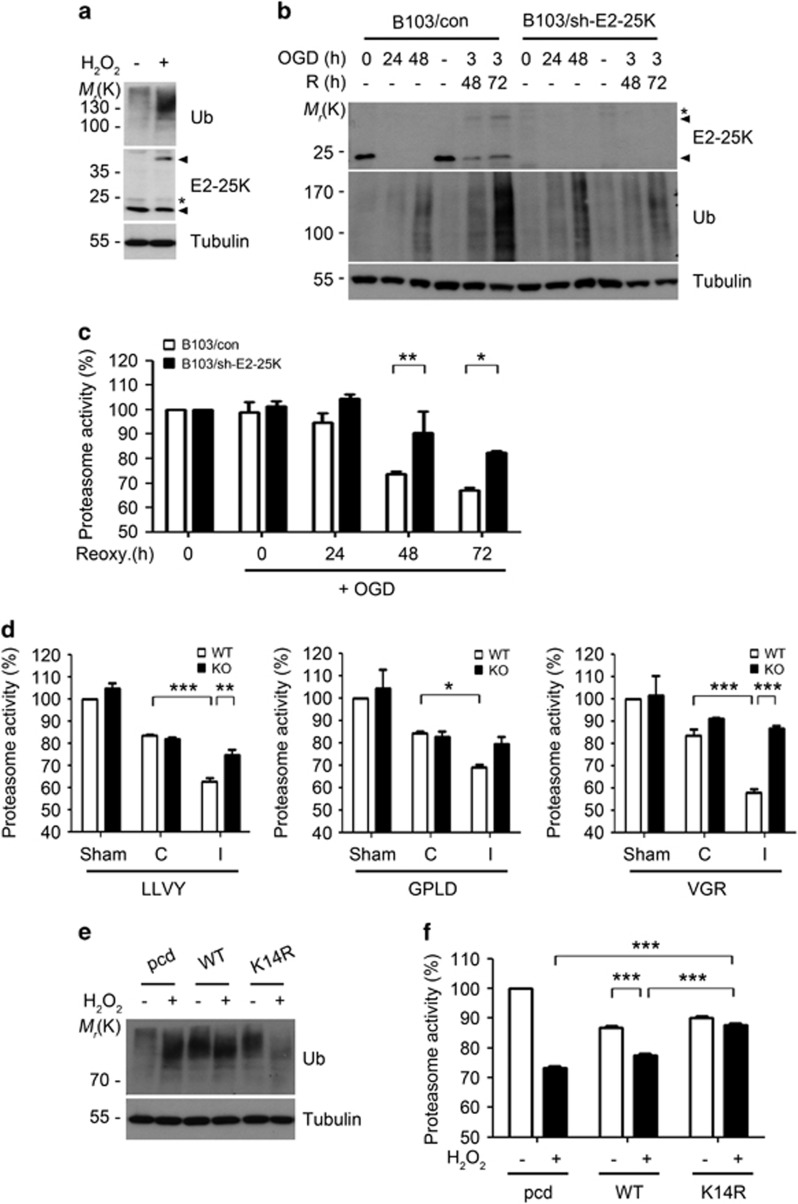
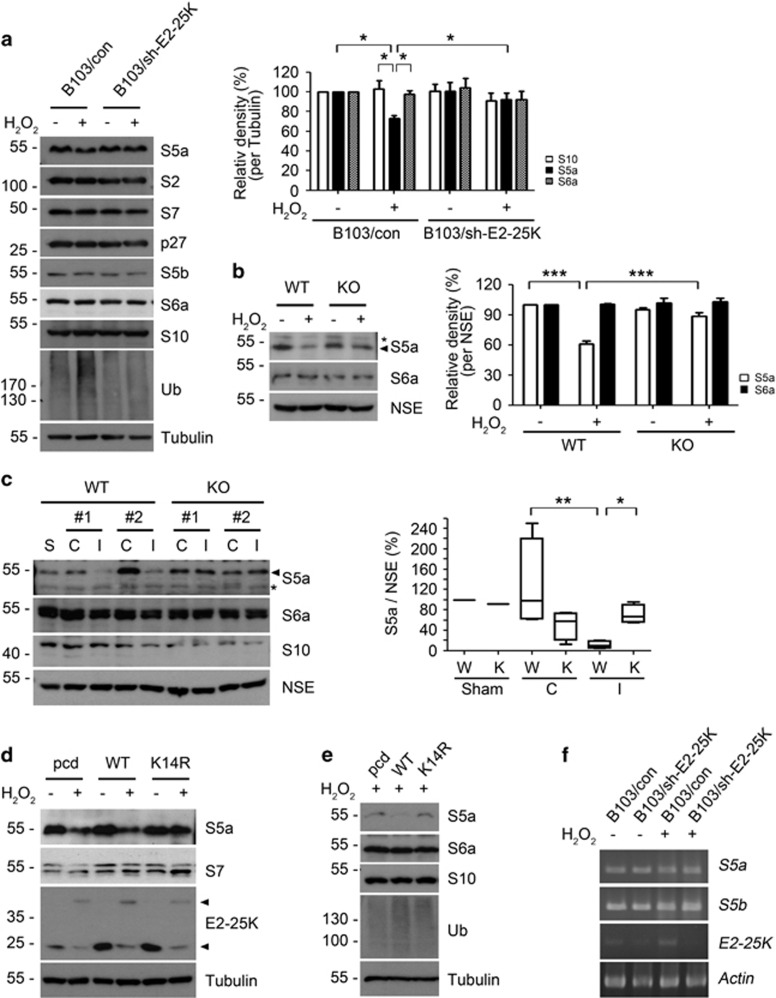
Cerebral ischemia/reperfusion (I/R) causes brain damage accompanied by ubiquitin accumulation and impairment of proteasome activity. In this study, we report that E2-25K, an E2-conjugating enzyme, is SUMOylated during oxidative stress and regulates cerebral I/R-induced damage. Knockdown of E2-25K expression protects against oxygen/glucose deprivation and reoxygenation (OGD/R)-induced neuronal cell death, whereas ectopic expression of E2-25K stimulates it. Compared with the control mice, cerebral infarction lesions and behavioral/neurological disorders are ameliorated in E2-25K knockout mice during middle cerebral artery occlusion and reperfusion. In particular, E2-25K is SUMOylated at Lys14 under oxidative stress, OGD/R and I/R to prompt cell death. Further, E2-25K downregulates the proteasome subunit S5a to impair proteasome complex and thus restrain proteasome activity under oxidative stress. This proteasome inhibitory activity of E2-25K is dependent on its SUMOylation. These results suggest that E2-25K has a crucial role in oxidative stress and cerebral I/R-induced damage through inhibiting proteasome via its SUMOylation.
脑缺血/再灌注(I/R)会导致脑损伤,并伴有泛素积累和蛋白酶体活性受损。在本研究中,我们报告了E2-25K(一种E2缀合酶)在氧化应激期间会发生SUMO化修饰,并调节脑I/R诱导的损伤。敲低E2-25K的表达可保护神经元免受氧/葡萄糖剥夺和复氧(OGD/R)诱导的细胞死亡,而E2-25K的异位表达则会促进这种细胞死亡。与对照小鼠相比,在大脑中动脉闭塞和再灌注期间,E2-25K基因敲除小鼠的脑梗死灶以及行为/神经功能障碍有所改善。特别地,在氧化应激、OGD/R和I/R条件下,E2-25K在赖氨酸14位点发生SUMO化修饰,从而促使细胞死亡。此外,E2-25K会下调蛋白酶体亚基S5a,以损害蛋白酶体复合物,进而在氧化应激下抑制蛋白酶体活性。E2-25K的这种蛋白酶体抑制活性依赖于其SUMO化修饰。这些结果表明,E2-25K通过其SUMO化修饰抑制蛋白酶体,在氧化应激和脑I/R诱导的损伤中起关键作用。