Blanco-Míguez Aitor, Meier-Kolthoff Jan P, Gutiérrez-Jácome Alberto, Göker Markus, Fdez-Riverola Florentino, Sánchez Borja, Lourenço Anália
ESEI-Department of Computer Science, University of Vigo, Edificio Politécnico, Campus Universitario As Lagoas s/n, Ourense, Spain.
Department of Microbiology and Biochemistry of Dairy Products, Instituto de Productos Lácteos de Asturias (IPLA), Consejo Superior de Investigaciones Científicas (CSIC), Villaviciosa, Asturias, Spain.
PLoS Comput Biol. 2016 Dec 29;12(12):e1005271. doi: 10.1371/journal.pcbi.1005271. eCollection 2016 Dec.
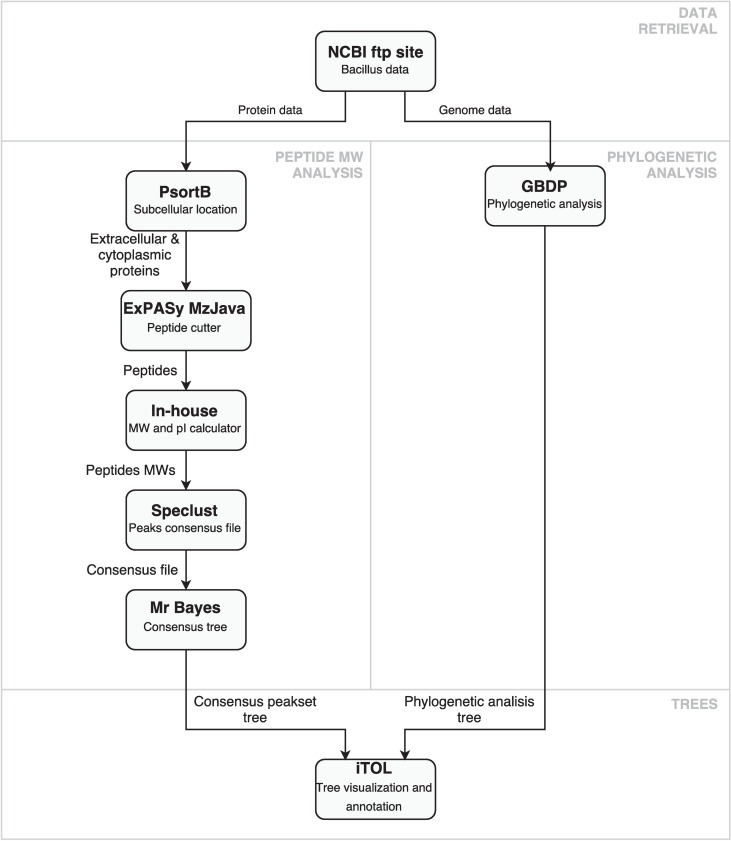
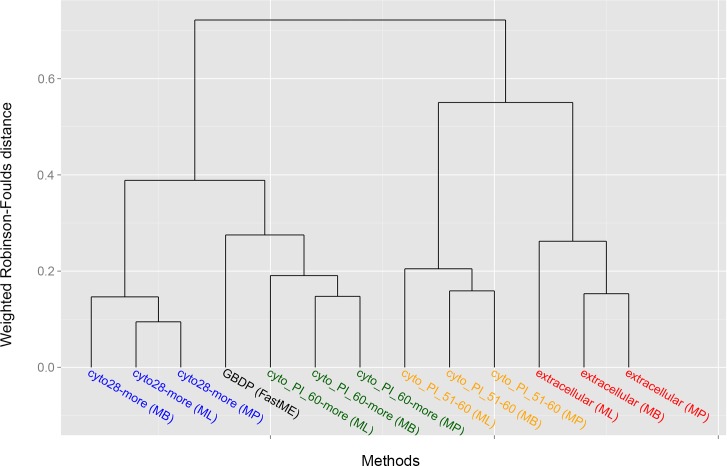
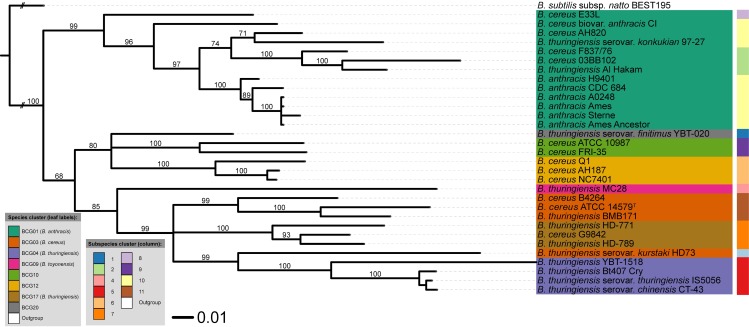
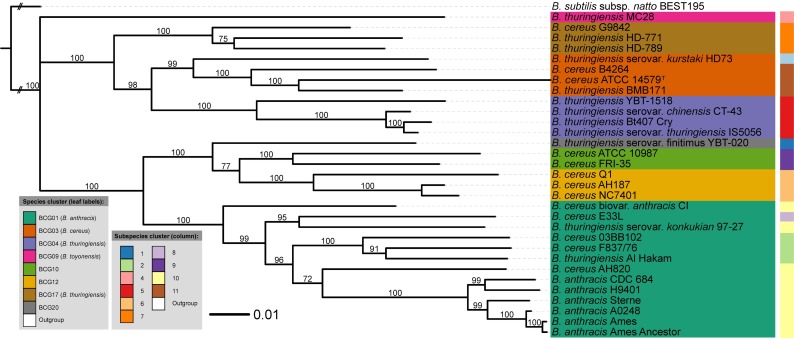
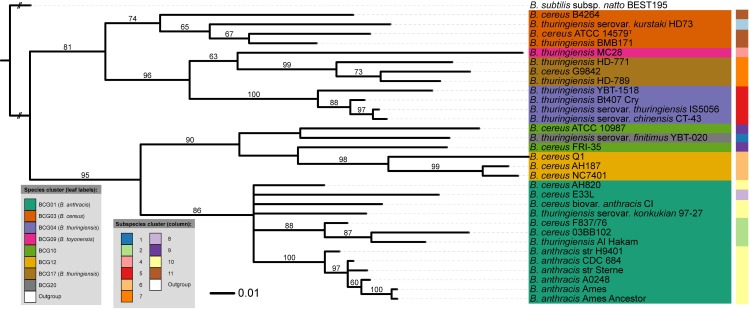
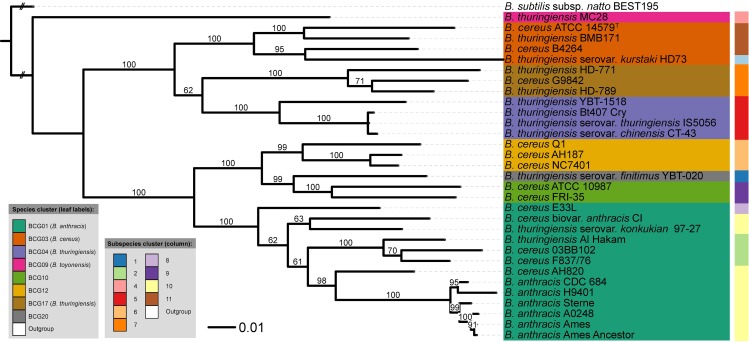
Typical bacterial strain differentiation methods are often challenged by high genetic similarity between strains. To address this problem, we introduce a novel in silico peptide fingerprinting method based on conventional wet-lab protocols that enables the identification of potential strain-specific peptides. These can be further investigated using in vitro approaches, laying a foundation for the development of biomarker detection and application-specific methods. This novel method aims at reducing large amounts of comparative peptide data to binary matrices while maintaining a high phylogenetic resolution. The underlying case study concerns the Bacillus cereus group, namely the differentiation of Bacillus thuringiensis, Bacillus anthracis and Bacillus cereus strains. Results show that trees based on cytoplasmic and extracellular peptidomes are only marginally in conflict with those based on whole proteomes, as inferred by the established Genome-BLAST Distance Phylogeny (GBDP) method. Hence, these results indicate that the two approaches can most likely be used complementarily even in other organismal groups. The obtained results confirm previous reports about the misclassification of many strains within the B. cereus group. Moreover, our method was able to separate the B. anthracis strains with high resolution, similarly to the GBDP results as benchmarked via Bayesian inference and both Maximum Likelihood and Maximum Parsimony. In addition to the presented phylogenomic applications, whole-peptide fingerprinting might also become a valuable complementary technique to digital DNA-DNA hybridization, notably for bacterial classification at the species and subspecies level in the future.
典型的细菌菌株鉴别方法常常受到菌株间高度遗传相似性的挑战。为解决这一问题,我们基于传统的湿实验室方案引入了一种新型的计算机模拟肽指纹图谱方法,该方法能够识别潜在的菌株特异性肽段。这些肽段可通过体外方法进一步研究,为生物标志物检测和特定应用方法的开发奠定基础。这种新方法旨在将大量的比较肽数据简化为二元矩阵,同时保持较高的系统发育分辨率。基础案例研究涉及蜡样芽孢杆菌组,即苏云金芽孢杆菌、炭疽芽孢杆菌和蜡样芽孢杆菌菌株的鉴别。结果表明,基于细胞质和细胞外肽组的树状图与基于全蛋白质组的树状图仅有轻微冲突,这是通过已确立的基因组- BLAST距离系统发育(GBDP)方法推断得出的。因此,这些结果表明,即使在其他生物类群中,这两种方法很可能也可互补使用。所得结果证实了先前关于蜡样芽孢杆菌组内许多菌株分类错误的报道。此外,我们的方法能够以高分辨率区分炭疽芽孢杆菌菌株,类似于通过贝叶斯推断以及最大似然法和最大简约法作为基准的GBDP结果。除了所展示的系统基因组学应用外,全肽指纹图谱未来可能还会成为数字DNA - DNA杂交的一种有价值的补充技术,特别是在物种和亚种水平的细菌分类方面。