Barman-Aksözen Jasmin, C Wiek Paulina, Bansode Vijay B, Koentgen Frank, Trüb Judith, Pelczar Pawel, Cinelli Paolo, Schneider-Yin Xiaoye, Schümperli Daniel, Minder Elisabeth I
Institute of Laboratory Medicine, Municipal Hospital Triemli, Zürich 8063, Switzerland.
Institute of Cell Biology, University of Bern, Bern 3012, Switzerland.
Dis Model Mech. 2017 Mar 1;10(3):225-233. doi: 10.1242/dmm.027755. Epub 2017 Jan 12.
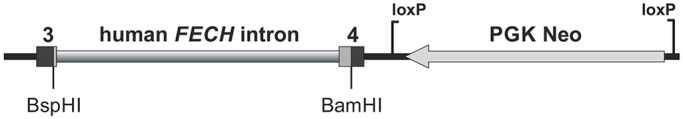
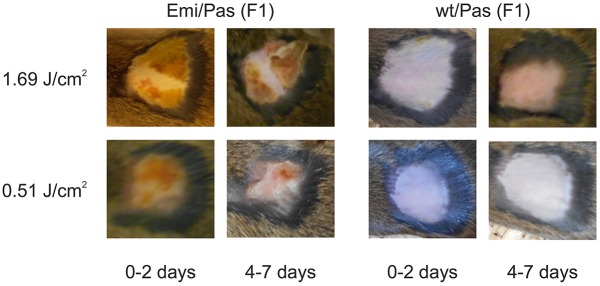
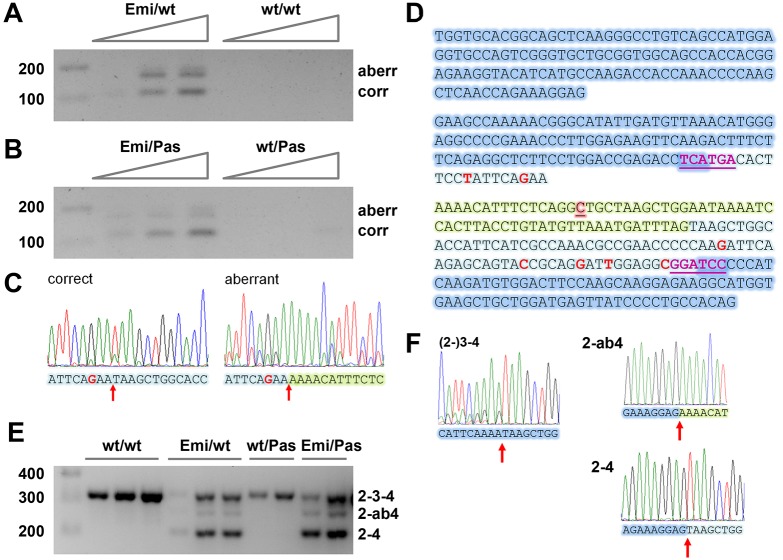
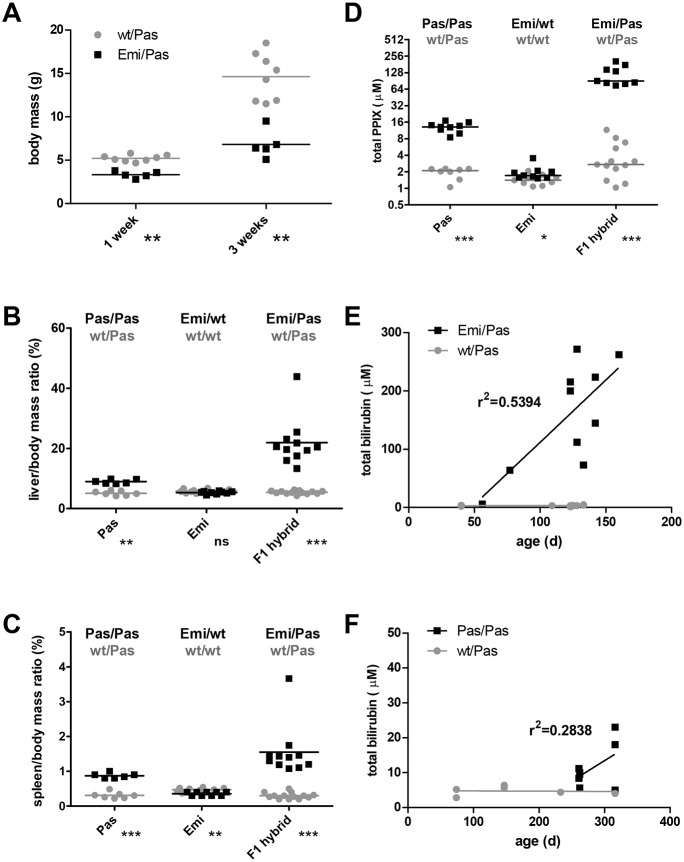
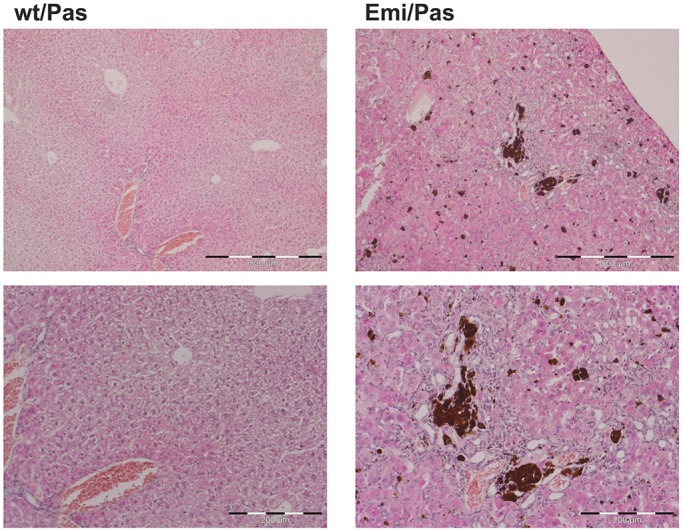
Erythropoietic protoporphyria (EPP) is caused by deficiency of ferrochelatase (FECH), which incorporates iron into protoporphyrin IX (PPIX) to form heme. Excitation of accumulated PPIX by light generates oxygen radicals that evoke excessive pain and, after longer light exposure, cause ulcerations in exposed skin areas of individuals with EPP. Moreover, ∼5% of the patients develop a liver dysfunction as a result of PPIX accumulation. Most patients (∼97%) have a severe mutation (Mut) to an intronic polymorphism (c.315-48C), which reduces ferrochelatase synthesis by stimulating the use of an aberrant 3' splice site 63 nt upstream of the normal site for exon 4. In contrast, with the predominant c.315-48T allele, the correct splice site is mostly used, and individuals with a T/Mut genotype do not develop EPP symptoms. Thus, the C allele is a potential target for therapeutic approaches that modify this splicing decision. To provide a model for pre-clinical studies of such approaches, we engineered a mouse containing a partly humanized gene with the c.315-48C polymorphism. F1 hybrids obtained by crossing these mice with another inbred line carrying a severe mutation (named m1Pas) show a very strong EPP phenotype that includes elevated PPIX in the blood, enlargement of liver and spleen, anemia, as well as strong pain reactions and skin lesions after a short period of light exposure. In addition to the expected use of the aberrant splice site, the mice also show a strong skipping of the partly humanized exon 3. This will limit the use of this model for certain applications and illustrates that engineering of a hybrid gene may have unforeseeable consequences on its splicing.
红细胞生成性原卟啉病(EPP)是由亚铁螯合酶(FECH)缺乏引起的,该酶将铁掺入原卟啉IX(PPIX)中以形成血红素。光激发积累的PPIX会产生活氧自由基,引发过度疼痛,并且在长时间光照后,会导致EPP患者暴露皮肤区域出现溃疡。此外,约5%的患者会因PPIX积累而出现肝功能障碍。大多数患者(约97%)存在严重突变(Mut),导致内含子多态性(c.315 - 48C),通过刺激使用比正常外显子4剪接位点上游63个核苷酸处的异常3'剪接位点,减少了亚铁螯合酶的合成。相比之下,对于占主导地位的c.315 - 48T等位基因,大多使用正确的剪接位点,具有T/Mut基因型的个体不会出现EPP症状。因此,C等位基因是改变这种剪接决定的治疗方法的潜在靶点。为了提供此类方法临床前研究的模型,我们构建了一只含有部分人源化基因且具有c.315 - 48C多态性的小鼠。将这些小鼠与另一个携带严重突变(命名为m1Pas)的近交系杂交获得的F1杂种表现出非常强烈的EPP表型,包括血液中PPIX升高、肝脏和脾脏肿大、贫血,以及在短时间光照后出现强烈的疼痛反应和皮肤损伤。除了预期使用异常剪接位点外,这些小鼠还表现出部分人源化的外显子3强烈跳跃。这将限制该模型在某些应用中的使用,并说明杂种基因工程可能对其剪接产生不可预见的后果。