Saito Kenichi, Kobayashi Eisuke, Yoshida Akihiko, Araki Yoshihiro, Kubota Daisuke, Tanzawa Yoshikazu, Kawai Akira, Yanagawa Takashi, Takagishi Kenji, Chuman Hirokazu
Division of Muscloskeletal Oncology, National Cancer Center Hospital, 5-5-1 Tsukiji, Chuo-ku, Tokyo, 104-0045, Japan.
Department of Orthopaedic Surgery, Gunma University Graduate School of Medicine, 3-39-22 Showa-machi, Maebashi, Gunma, 371-8511, Japan.
BMC Musculoskelet Disord. 2017 Jan 23;18(1):31. doi: 10.1186/s12891-017-1390-y.
Angiomatoid fibrous histiocytoma (AFH) is a rare soft tissue tumor of intermediate biologic potential. Because of its rarity and nonspecific radiological and diverse pathological findings, AFH is often clinically misdiagnosed. However, few clinical reports have described this tumor. As reported herein, we analyzed the clinical and radiological features and clinical outcomes of AFH.
We retrospectively reviewed the medical records of seven cases histopathologically diagnosed as AFH. We examined clinical features, MRI findings, histopathological diagnoses, treatments, and outcomes.
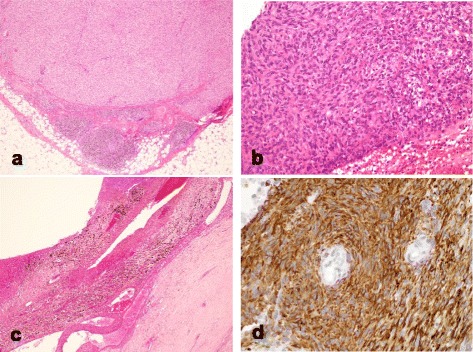
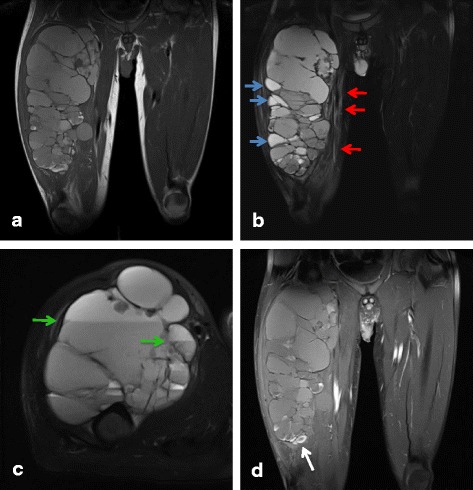
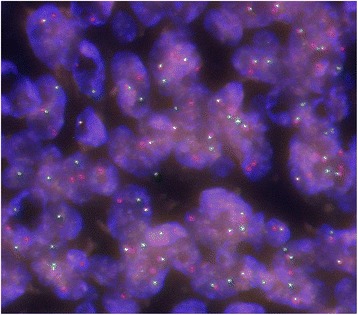
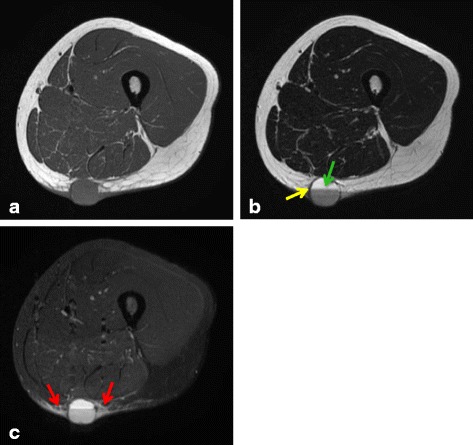
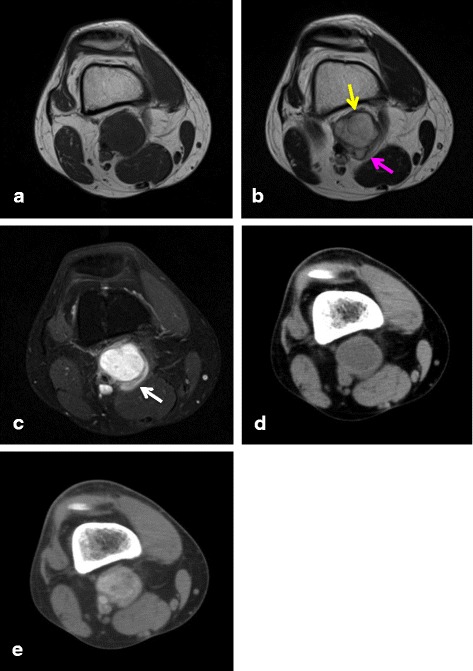
These seven cases comprised five male and two female patients with ages ranging from 8 to 50 years old. The primary locations included upper extremities in 2, lower extremities in 4, and the inguinal region in one patient. Of the tumors, 4 occurred in subcutaneous tissues and 3 occurred in deep tissues. No cases were diagnosed as AFH from MRI and needle biopsy results. All cases were diagnosed histopathologically after excision. After treatment, 2 patients (29%) had tumor recurrence and metastasis, one of whom died from disease progression. These 2 aggressive cases involved both EWSR1 and CREB1 gene rearrangements as determined by FISH. The other patients were alive and well without recurrence or metastasis.
AFH is a rare tumor that is difficult to diagnose. Therefore, it tends to be misdiagnosed and to be treated inadequately by referring physicians. Surgeons must therefore be mindful of the presence of AFH, learn about appropriate treatment necessary for this tumor, and conduct careful follow-up because AFH can engender poor outcomes.
血管样纤维组织细胞瘤(AFH)是一种具有中等生物学潜能的罕见软组织肿瘤。由于其罕见性以及非特异性的影像学表现和多样的病理结果,AFH在临床上常被误诊。然而,关于这种肿瘤的临床报道很少。正如本文所报道的,我们分析了AFH的临床、影像学特征及临床结局。
我们回顾性分析了7例经组织病理学诊断为AFH的病例的病历资料。我们检查了临床特征、MRI表现、组织病理学诊断、治疗方法及结局。
这7例患者中,男性5例,女性2例,年龄在8至50岁之间。原发部位包括上肢2例,下肢4例,腹股沟区1例。其中4例肿瘤位于皮下组织,3例位于深部组织。MRI和针吸活检结果均未诊断出AFH。所有病例均在切除术后经组织病理学确诊。治疗后,2例患者(29%)出现肿瘤复发和转移,其中1例死于疾病进展。通过荧光原位杂交(FISH)检测确定,这2例侵袭性病例均涉及EWSR1和CREB1基因重排。其他患者均存活且情况良好,无复发或转移。
AFH是一种罕见的难以诊断的肿瘤。因此,转诊医生往往容易误诊并给予不充分的治疗。外科医生必须因此留意AFH的存在,了解针对该肿瘤所需的适当治疗方法,并进行仔细随访,因为AFH可能导致不良结局。