Li Miaoxin, Li Jiang, Li Mulin Jun, Pan Zhicheng, Hsu Jacob Shujui, Liu Dajiang J, Zhan Xiaowei, Wang Junwen, Song Youqiang, Sham Pak Chung
Department of Medical Genetics, Center for Genome Research, Center for Precision Medicine, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, 510080, China.
The Centre for Genomic Sciences, the University of Hong Kong, Pokfulam, Hong Kong.
Nucleic Acids Res. 2017 May 19;45(9):e75. doi: 10.1093/nar/gkx019.
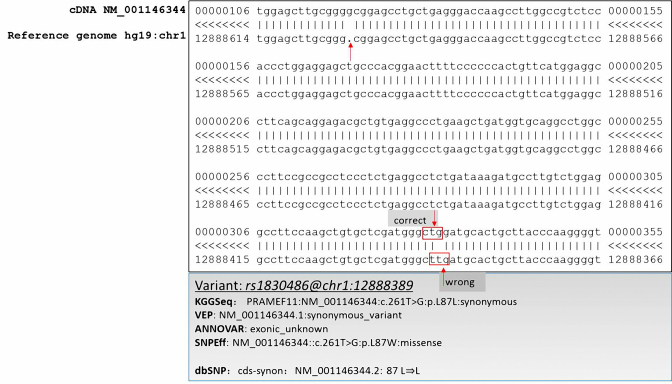
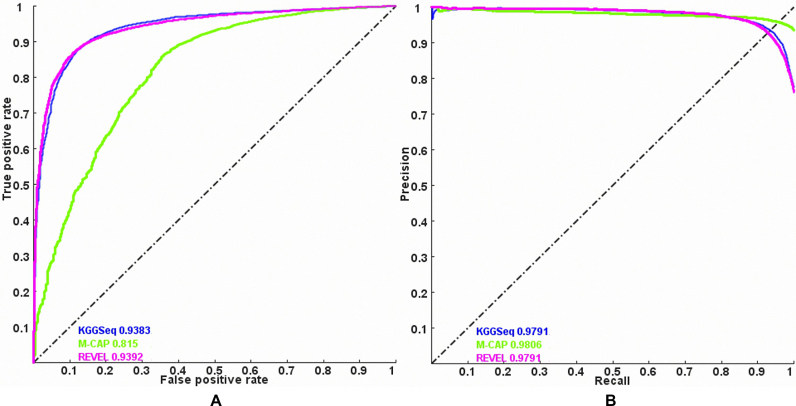
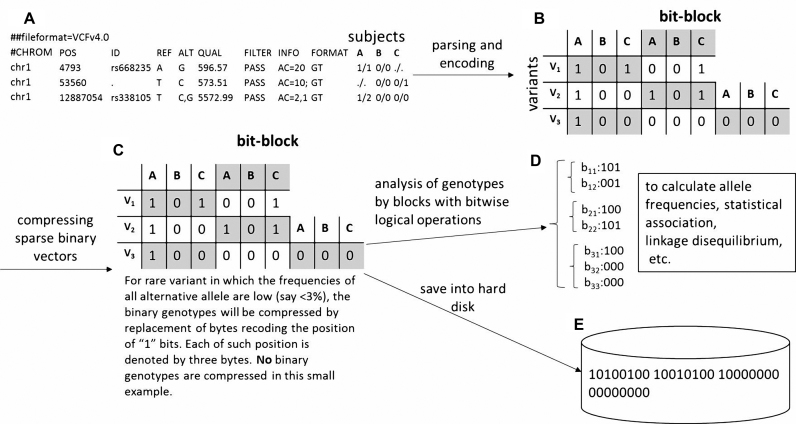
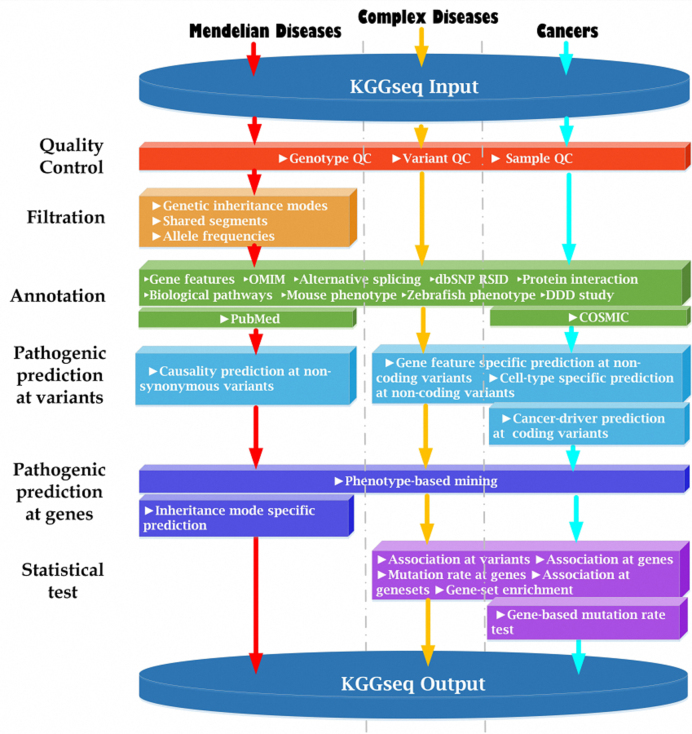
Whole genome sequencing (WGS) is a promising strategy to unravel variants or genes responsible for human diseases and traits. However, there is a lack of robust platforms for a comprehensive downstream analysis. In the present study, we first proposed three novel algorithms, sequence gap-filled gene feature annotation, bit-block encoded genotypes and sectional fast access to text lines to address three fundamental problems. The three algorithms then formed the infrastructure of a robust parallel computing framework, KGGSeq, for integrating downstream analysis functions for whole genome sequencing data. KGGSeq has been equipped with a comprehensive set of analysis functions for quality control, filtration, annotation, pathogenic prediction and statistical tests. In the tests with whole genome sequencing data from 1000 Genomes Project, KGGSeq annotated several thousand more reliable non-synonymous variants than other widely used tools (e.g. ANNOVAR and SNPEff). It took only around half an hour on a small server with 10 CPUs to access genotypes of ∼60 million variants of 2504 subjects, while a popular alternative tool required around one day. KGGSeq's bit-block genotype format used 1.5% or less space to flexibly represent phased or unphased genotypes with multiple alleles and achieved a speed of over 1000 times faster to calculate genotypic correlation.
全基因组测序(WGS)是一种很有前景的策略,可用于揭示导致人类疾病和性状的变异或基因。然而,目前缺乏强大的平台来进行全面的下游分析。在本研究中,我们首先提出了三种新颖的算法,即序列缺口填充基因特征注释、位块编码基因型和文本行的分段快速访问,以解决三个基本问题。这三种算法随后构成了一个强大的并行计算框架KGGSeq的基础架构,用于整合全基因组测序数据的下游分析功能。KGGSeq配备了一套全面的分析功能,用于质量控制、过滤、注释、致病性预测和统计测试。在对来自千人基因组计划的全基因组测序数据进行测试时,KGGSeq注释的可靠非同义变异比其他广泛使用的工具(如ANNOVAR和SNPEff)多出数千个。在一台配备10个CPU的小型服务器上,只需大约半小时就能获取2504名受试者约6000万个变异的基因型,而另一个常用工具则需要大约一天时间。KGGSeq的位块基因型格式使用的空间不到1.5%,能够灵活地表示具有多个等位基因的分阶段或未分阶段基因型,并且计算基因型相关性的速度快了1000倍以上。