Farmanbar Amir, Firouzi Sanaz, Park Sung-Joon, Nakai Kenta, Uchimaru Kaoru, Watanabe Toshiki
Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Tokyo, Japan.
Laboratory of Functional Analysis in silico, Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo, Japan.
BMC Med Genomics. 2017 Jan 31;10(1):4. doi: 10.1186/s12920-016-0241-2.
Clonal expansion of leukemic cells leads to onset of adult T-cell leukemia (ATL), an aggressive lymphoid malignancy with a very poor prognosis. Infection with human T-cell leukemia virus type-1 (HTLV-1) is the direct cause of ATL onset, and integration of HTLV-1 into the human genome is essential for clonal expansion of leukemic cells. Therefore, monitoring clonal expansion of HTLV-1-infected cells via isolation of integration sites assists in analyzing infected individuals from early infection to the final stage of ATL development. However, because of the complex nature of clonal expansion, the underlying mechanisms have yet to be clarified. Combining computational/mathematical modeling with experimental and clinical data of integration site-based clonality analysis derived from next generation sequencing technologies provides an appropriate strategy to achieve a better understanding of ATL development.
As a comprehensively interdisciplinary project, this study combined three main aspects: wet laboratory experiments, in silico analysis and empirical modeling.
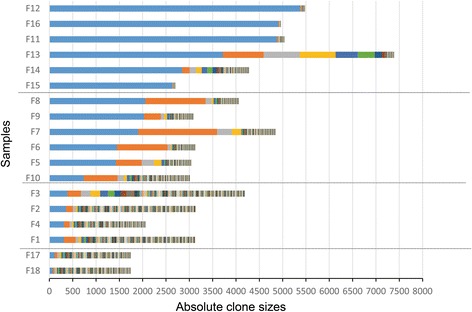
We analyzed clinical samples from HTLV-1-infected individuals with a broad range of proviral loads using a high-throughput methodology that enables isolation of HTLV-1 integration sites and accurate measurement of the size of infected clones. We categorized clones into four size groups, "very small", "small", "big", and "very big", based on the patterns of clonal growth and observed clone sizes. We propose an empirical formal model based on deterministic finite state automata (DFA) analysis of real clinical samples to illustrate patterns of clonal expansion.
Through the developed model, we have translated biological data of clonal expansion into the formal language of mathematics and represented the observed clonality data with DFA. Our data suggest that combining experimental data (absolute size of clones) with DFA can describe the clonality status of patients. This kind of modeling provides a basic understanding as well as a unique perspective for clarifying the mechanisms of clonal expansion in ATL.
白血病细胞的克隆性扩增导致成人T细胞白血病(ATL)的发生,这是一种侵袭性淋巴细胞恶性肿瘤,预后极差。人类T细胞白血病病毒1型(HTLV-1)感染是ATL发病的直接原因,HTLV-1整合到人类基因组中对于白血病细胞的克隆性扩增至关重要。因此,通过分离整合位点来监测HTLV-1感染细胞的克隆性扩增有助于分析感染个体从早期感染到ATL发展的最终阶段。然而,由于克隆性扩增的复杂性,其潜在机制尚未阐明。将计算/数学建模与来自下一代测序技术的基于整合位点的克隆性分析的实验和临床数据相结合,为更好地理解ATL发展提供了一种合适的策略。
作为一个全面的跨学科项目,本研究结合了三个主要方面:湿实验室实验、计算机分析和实证建模。
我们使用一种高通量方法分析了来自HTLV-1感染个体的临床样本,这些样本具有广泛的前病毒载量,该方法能够分离HTLV-1整合位点并准确测量感染克隆的大小。我们根据克隆生长模式和观察到的克隆大小将克隆分为四个大小组,即“非常小”、“小”、“大”和“非常大”。我们基于对真实临床样本的确定性有限状态自动机(DFA)分析提出了一个实证形式模型,以说明克隆性扩增模式。
通过所开发的模型,我们已将克隆性扩增的生物学数据转化为数学形式语言,并用DFA表示观察到的克隆性数据。我们的数据表明,将实验数据(克隆的绝对大小)与DFA相结合可以描述患者的克隆性状态。这种建模为阐明ATL中克隆性扩增机制提供了基本理解以及独特视角。