Way Gregory P, Allaway Robert J, Bouley Stephanie J, Fadul Camilo E, Sanchez Yolanda, Greene Casey S
Genomics and Computational Biology Graduate Program, University of Pennsylvania, Philadelphia, PA, USA.
Department of Systems Pharmacology and Translational Therapeutics, University of Pennsylvania, 10-131 SCTR 34th and Civic Center Blvd, Philadelphia, PA, 19104, USA.
BMC Genomics. 2017 Feb 6;18(1):127. doi: 10.1186/s12864-017-3519-7.
We have identified molecules that exhibit synthetic lethality in cells with loss of the neurofibromin 1 (NF1) tumor suppressor gene. However, recognizing tumors that have inactivation of the NF1 tumor suppressor function is challenging because the loss may occur via mechanisms that do not involve mutation of the genomic locus. Degradation of the NF1 protein, independent of NF1 mutation status, phenocopies inactivating mutations to drive tumors in human glioma cell lines. NF1 inactivation may alter the transcriptional landscape of a tumor and allow a machine learning classifier to detect which tumors will benefit from synthetic lethal molecules.
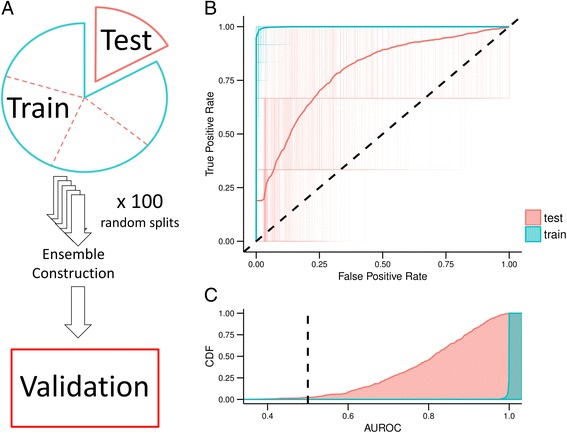
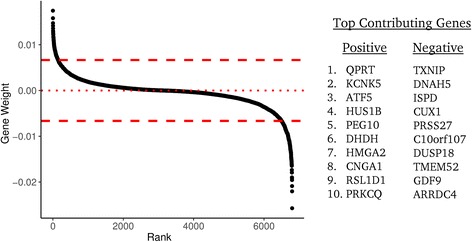
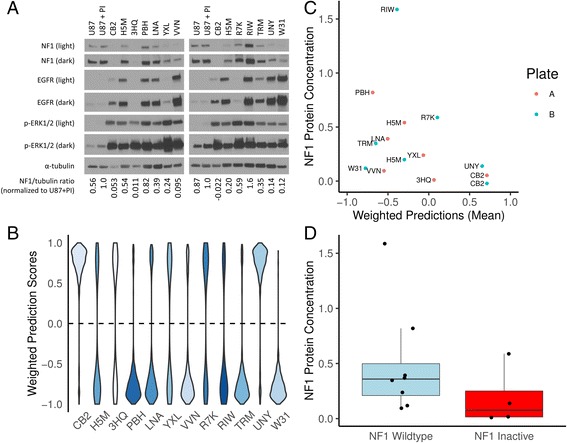
We developed a strategy to predict tumors with low NF1 activity and hence tumors that may respond to treatments that target cells lacking NF1. Using RNAseq data from The Cancer Genome Atlas (TCGA), we trained an ensemble of 500 logistic regression classifiers that integrates mutation status with whole transcriptomes to predict NF1 inactivation in glioblastoma (GBM). On TCGA data, the classifier detected NF1 mutated tumors (test set area under the receiver operating characteristic curve (AUROC) mean = 0.77, 95% quantile = 0.53 - 0.95) over 50 random initializations. On RNA-Seq data transformed into the space of gene expression microarrays, this method produced a classifier with similar performance (test set AUROC mean = 0.77, 95% quantile = 0.53 - 0.96). We applied our ensemble classifier trained on the transformed TCGA data to a microarray validation set of 12 samples with matched RNA and NF1 protein-level measurements. The classifier's NF1 score was associated with NF1 protein concentration in these samples.
We demonstrate that TCGA can be used to train accurate predictors of NF1 inactivation in GBM. The ensemble classifier performed well for samples with very high or very low NF1 protein concentrations but had mixed performance in samples with intermediate NF1 concentrations. Nevertheless, high-performing and validated predictors have the potential to be paired with targeted therapies and personalized medicine.
我们已经鉴定出在神经纤维瘤病1型(NF1)肿瘤抑制基因缺失的细胞中表现出合成致死性的分子。然而,识别具有NF1肿瘤抑制功能失活的肿瘤具有挑战性,因为这种缺失可能通过不涉及基因组位点突变的机制发生。NF1蛋白的降解,与NF1突变状态无关,模拟了失活突变,从而在人胶质瘤细胞系中驱动肿瘤发生。NF1失活可能会改变肿瘤的转录图谱,并使机器学习分类器能够检测出哪些肿瘤将从合成致死分子中获益。
我们开发了一种策略来预测NF1活性低的肿瘤,从而预测可能对靶向缺乏NF1的细胞的治疗产生反应的肿瘤。利用来自癌症基因组图谱(TCGA)的RNAseq数据,我们训练了一个由500个逻辑回归分类器组成的集成模型,该模型将突变状态与整个转录组相结合,以预测胶质母细胞瘤(GBM)中的NF1失活。在TCGA数据上,经过50次随机初始化后,该分类器检测到NF1突变肿瘤(测试集受试者操作特征曲线下面积(AUROC)均值 = 0.77,95%分位数 = 0.53 - 0.95)。在转换为基因表达微阵列空间的RNA-Seq数据上,该方法产生了一个性能相似的分类器(测试集AUROC均值 = 0.77,95%分位数 = 0.53 - 0.96)。我们将在转换后的TCGA数据上训练的集成分类器应用于一个包含12个样本的微阵列验证集,这些样本具有匹配的RNA和NF1蛋白水平测量值。该分类器的NF1评分与这些样本中的NF1蛋白浓度相关。
我们证明TCGA可用于训练GBM中NF1失活的准确预测指标。该集成分类器在NF1蛋白浓度非常高或非常低的样本中表现良好,但在NF1浓度中等的样本中表现不一。尽管如此,高性能且经过验证的预测指标有可能与靶向治疗和个性化医疗相结合。