NanoString Technologies, Inc., 530 Fairview Ave N, Seattle, WA 98109 USA.
Tumor Vaccine Group, University of Washington, 850 Republican Street, Box 358050, Seattle, WA 98109 USA.
J Immunother Cancer. 2017 Feb 21;5:18. doi: 10.1186/s40425-017-0215-8. eCollection 2017.
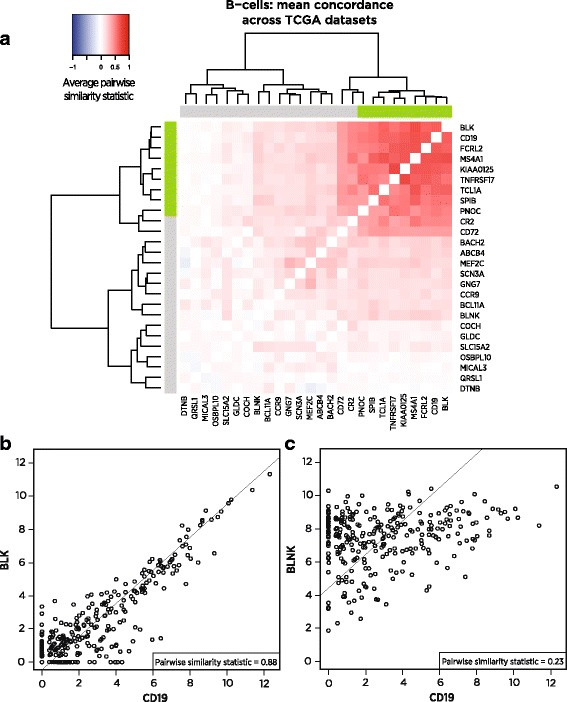
Assays of the abundance of immune cell populations in the tumor microenvironment promise to inform immune oncology research and the choice of immunotherapy for individual patients. We propose to measure the intratumoral abundance of various immune cell populations with gene expression. In contrast to IHC and flow cytometry, gene expression assays yield high information content from a clinically practical workflow. Previous studies of gene expression in purified immune cells have reported hundreds of genes showing enrichment in a single cell type, but the utility of these genes in tumor samples is unknown. We use co-expression patterns in large tumor gene expression datasets to evaluate previously reported candidate cell type marker genes lists, eliminate numerous false positives and identify a subset of high confidence marker genes.
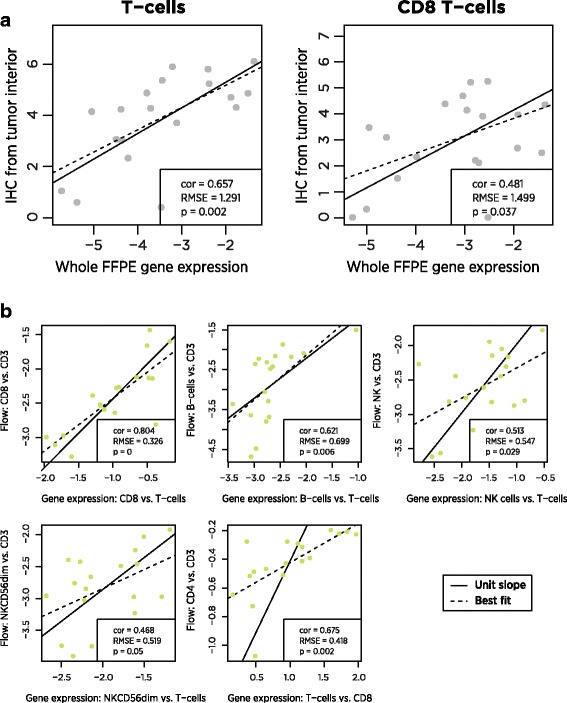
Using a novel statistical tool, we use co-expression patterns in 9986 samples from The Cancer Genome Atlas (TCGA) to evaluate previously reported cell type marker genes. We compare immune cell scores derived from these genes to measurements from flow cytometry and immunohistochemistry. We characterize the reproducibility of our cell scores in replicate runs of RNA extracted from FFPE tumor tissue.
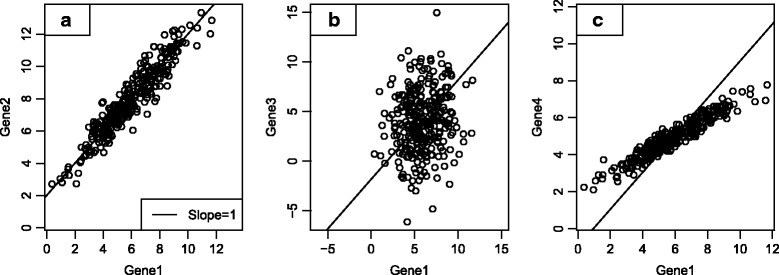
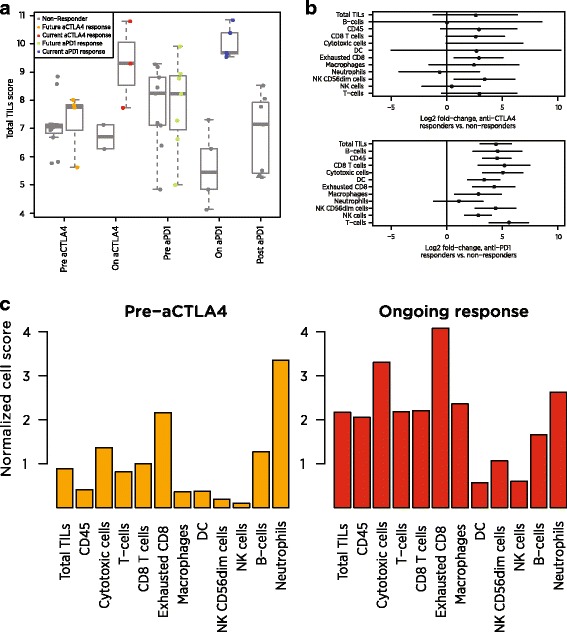
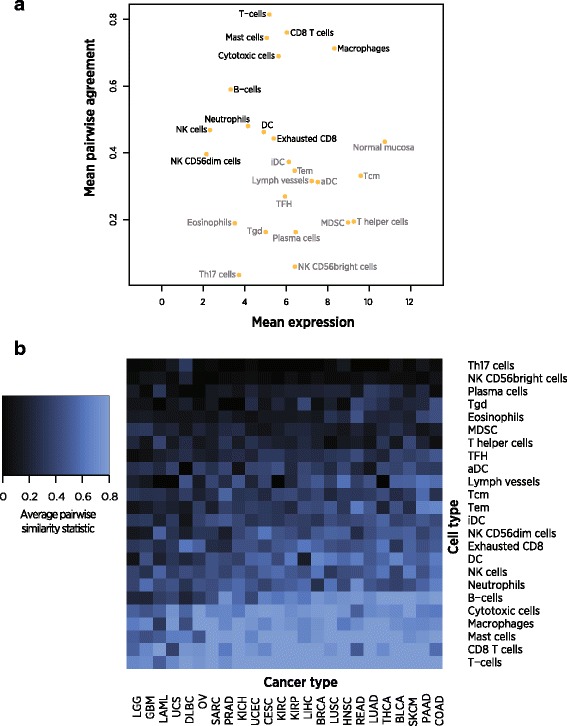
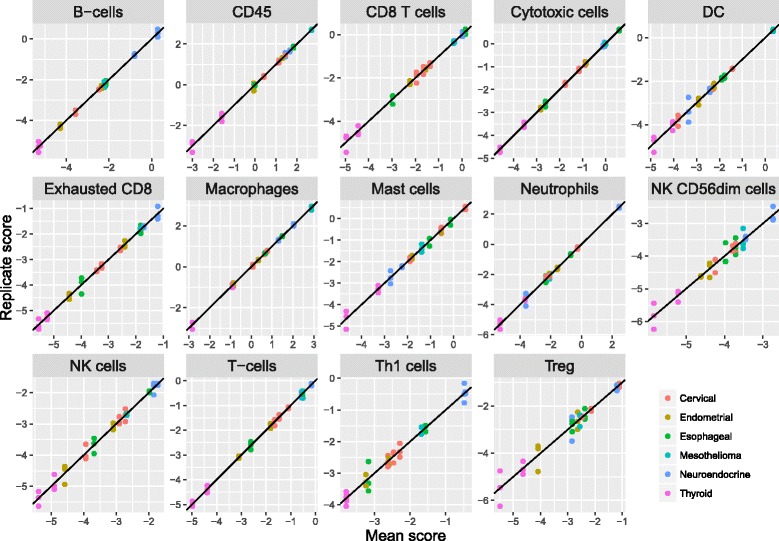
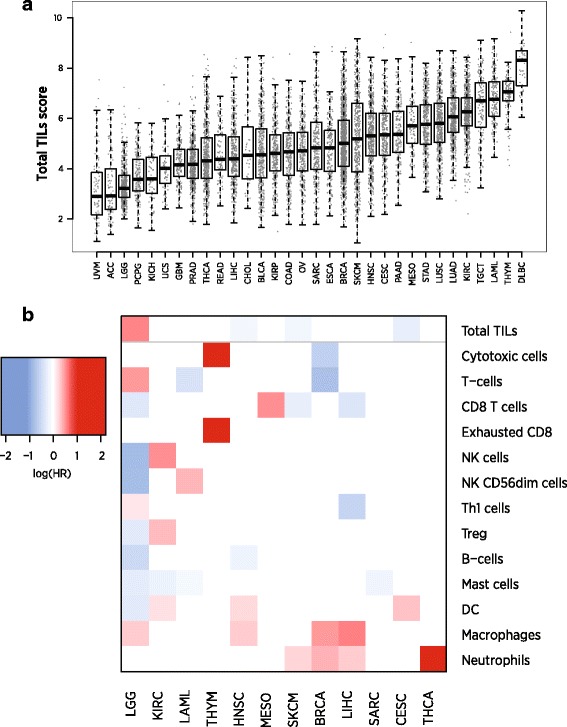
We identify a list of 60 marker genes whose expression levels measure 14 immune cell populations. Cell type scores calculated from these genes are concordant with flow cytometry and IHC readings, show high reproducibility in replicate RNA samples from FFPE tissue and enable detailed analyses of the anti-tumor immune response in TCGA. In an immunotherapy dataset, they separate responders and non-responders early on therapy and provide an intricate picture of the effects of checkpoint inhibition. Most genes previously reported to be enriched in a single cell type have co-expression patterns inconsistent with cell type specificity.
Due to their concise gene set, computational simplicity and utility in tumor samples, these cell type gene signatures may be useful in future discovery research and clinical trials to understand how tumors and therapeutic intervention shape the immune response.
在肿瘤微环境中检测免疫细胞群体的丰度有望为免疫肿瘤学研究和为个体患者选择免疫疗法提供信息。我们提议使用基因表达来测量肿瘤内各种免疫细胞群体的丰度。与 IHC 和流式细胞术相比,基因表达测定从临床实用的工作流程中获得了高信息量。以前对纯化免疫细胞中的基因表达的研究报告了数百个在单个细胞类型中富集的基因,但这些基因在肿瘤样本中的用途尚不清楚。我们使用大型肿瘤基因表达数据集的共表达模式来评估先前报道的候选细胞类型标记基因列表,消除大量假阳性并确定一组高可信度的标记基因。
使用一种新的统计工具,我们使用来自癌症基因组图谱 (TCGA) 的 9986 个样本中的共表达模式来评估先前报道的细胞类型标记基因。我们将这些基因衍生的免疫细胞评分与流式细胞术和免疫组织化学测量结果进行比较。我们描述了从 FFPE 肿瘤组织中提取的 RNA 重复运行中细胞评分的可重复性。
我们确定了一组 60 个标记基因,其表达水平可测量 14 种免疫细胞群体。从这些基因计算的细胞分数与流式细胞术和 IHC 读数一致,在 FFPE 组织的重复 RNA 样本中具有高重复性,并能够在 TCGA 中对肿瘤的抗肿瘤免疫反应进行详细分析。在免疫治疗数据集,它们在治疗早期将应答者和无应答者分开,并提供了检查点抑制作用的复杂图像。以前报道的在单个细胞类型中富集的大多数基因的共表达模式与细胞类型特异性不一致。
由于其简洁的基因集、计算简单性以及在肿瘤样本中的实用性,这些细胞类型基因特征可能在未来的发现研究和临床试验中有用,以了解肿瘤和治疗干预如何塑造免疫反应。