Bolognini Ramona, Gerth-Kahlert Christina, Abegg Mathias, Bartholdi Deborah, Mathis Nicolas, Sturm Veit, Gallati Sabina, Schaller André
Division of Human Genetics, Department of Pediatrics, Inselspital, Bern University Hospital, University of Bern, Freiburgstrasse 15, 3010, Bern, Switzerland.
Graduate School for Cellular and Biomedical Sciences (GCB), University of Bern, Bern, Switzerland.
BMC Med Genet. 2017 Feb 28;18(1):22. doi: 10.1186/s12881-017-0383-x.
We report two novel splice region mutations in OPA1 in two unrelated families presenting with autosomal-dominant optic atrophy type 1 (ADOA1) (ADOA or Kjer type optic atrophy). Mutations in OPA1 encoding a mitochondrial inner membrane protein are a major cause of ADOA.
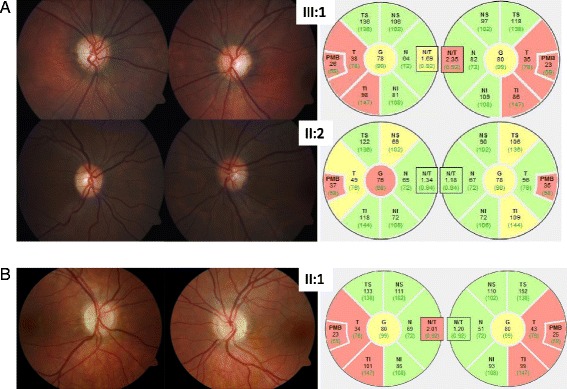
We analyzed two unrelated families including four affected individuals clinically suspicious of ADOA. Standard ocular examinations were performed in affected individuals of both families. All coding exons, as well as exon-intron boundaries of the OPA1 gene were sequenced. In addition, multiplex ligation-dependent probe amplification (MLPA) was performed to uncover copy number variations in OPA1. mRNA processing was monitored using RT-PCR and subsequent cDNA analysis.
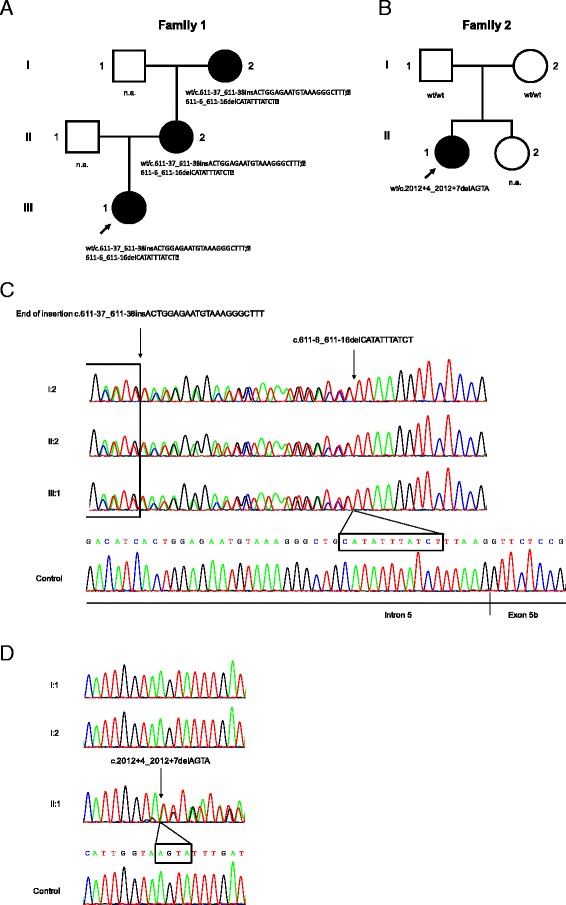
We report two novel splice region mutations in OPA1 in two unrelated individuals and their affected relatives, which were previously not described in the literature. In one family the heterozygous insertion and deletion c.[611-37_611-38insACTGGAGAATGTAAAGGGCTTT;611-6_611-16delCATATTTATCT] was found in all investigated family members leading to the activation of an intronic cryptic splice site. In the second family sequencing of OPA1 disclosed a de novo heterozygous deletion c.2012+4_2012+7delAGTA resulting in exon 18 and 19 skipping, which was not detected in healthy family members.
We identified two novel intronic mutations in OPA1 affecting the correct OPA1 pre-mRNA splicing, which was confirmed by OPA1 cDNA analysis. This study shows the importance of transcript analysis to determine the consequences of unclear intronic mutations in OPA1 in proximity to the intron-exon boundaries.
我们报告了两个不相关家族中OPA1基因的两个新的剪接区域突变,这两个家族均表现为常染色体显性遗传性1型视神经萎缩(ADOA1)(ADOA或凯尔型视神经萎缩)。编码线粒体内膜蛋白的OPA1基因突变是ADOA的主要病因。
我们分析了两个不相关的家族,其中包括四名临床上疑似患有ADOA的患者。对两个家族的患病个体进行了标准眼科检查。对OPA1基因的所有编码外显子以及外显子-内含子边界进行了测序。此外,进行了多重连接依赖探针扩增(MLPA)以检测OPA1基因的拷贝数变异。使用逆转录聚合酶链反应(RT-PCR)和后续的cDNA分析监测mRNA加工过程。
我们在两个不相关个体及其患病亲属中报告了OPA1基因的两个新的剪接区域突变,这些突变以前在文献中未曾描述。在一个家族中,所有被调查的家族成员均发现杂合插入和缺失c.[611-37_611-38insACTGGAGAATGTAAAGGGCTTT;611-6_611-16delCATATTTATCT],导致一个内含子隐蔽剪接位点被激活。在第二个家族中,OPA1基因测序发现一个新生杂合缺失c.2012+4_2012+7delAGTA,导致外显子18和19跳跃,在健康家族成员中未检测到。
我们在OPA1基因中鉴定出两个新的内含子突变,影响了OPA1前体mRNA的正确剪接,这一点通过OPA1 cDNA分析得到证实。本研究表明转录本分析对于确定OPA1基因内含子-外显子边界附近不明内含子突变的后果具有重要意义。