Muñoz-Bonet Juan Ignacio, Ortega-Sánchez María Del Carmen, León Guijarro José Luis
Pediatric Intensive Care Unit, Hospital Clínico Universitario, University of Valencia, Av. Blasco Ibáñez 17, 46010, Valencia, Spain.
Department of Pediatrics, Hospital Clínico Universitario, Av. Blasco Ibáñez 17, 46010, Valencia, Spain.
Ital J Pediatr. 2017 Jan 19;43(1):12. doi: 10.1186/s13052-017-0333-4.
3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) lyase deficiency is a rare inborn error of metabolism characterized by recurrent metabolic crises caused by fasting, intercurrent illness and excessive physical exercise. Non ketotic hypoglycemia is normally the cause of primary symptoms but without an immediate treatment the illness can evolve into a worsening metabolic state resembling the Reye's syndrome that may cause the patient's death. We report a case with some clinical and therapeutic features not previously described.
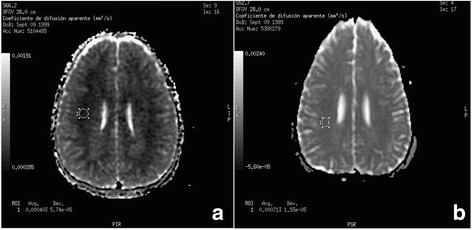
Patient with HMG-CoA lyase deficiency whom after diagnosis at 2 years of age was re-admitted 12 years later, after severe metabolic decompensation following consumption of alcohol. Despite a quick correction of hypoglycemia, within the following few hours, the patient fell into a coma. Suspecting intracranial hypertension (ICH), the patient required mechanical ventilation. Although liver cytolysis was minimal, hyperamoniemia reached 1394 μmol/L, returning to normal, a few hours after administering sodium phenylacetate and sodium benzoate, whose use has not been reported in these patients. Brain edema was evidenced in the computed tomography and by the magnetic resonance imaging that determined that the edema was cytotoxic, as quantified with the restriction of diffusion in the apparent diffusion coefficient map. During the recovery of the ICH, we belatedly, detected vasospasm moderate-severe that was treated with nimodipine. Currently, the patient maintains clinical normality.
The alcohol consumption must be avoided in patients with HMG-CoA lyase deficiency. In our patient hyperamoniemia was effectively treated with sodium phenylacetate and sodium benzoate. Magnetic resonance imaging showed and quantified the cytotoxic brain edema. Belatedly, a cerebral vasospasm was an additional mechanism of cerebral injury. None of these observations has been previously reported.
3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)裂解酶缺乏症是一种罕见的先天性代谢紊乱疾病,其特征为禁食、并发疾病及过度体育锻炼引发反复代谢危机。非酮症低血糖通常是主要症状的病因,但如不及时治疗,病情可能演变为类似瑞氏综合征的恶化代谢状态,进而导致患者死亡。我们报告一例具有一些此前未描述的临床及治疗特征的病例。
一名患有HMG-CoA裂解酶缺乏症的患者,2岁时确诊,12年后因饮酒后严重代谢失代偿再次入院。尽管低血糖迅速得到纠正,但在接下来的几个小时内,患者陷入昏迷。怀疑有颅内高压(ICH),患者需要机械通气。尽管肝细胞溶解轻微,但高氨血症达到1394μmol/L,在给予苯乙酸钠和苯甲酸钠后数小时恢复正常,此前这些患者中尚未有使用这两种药物的报道。计算机断层扫描和磁共振成像证实存在脑水肿,并确定该水肿为细胞毒性水肿,通过表观扩散系数图中的扩散受限进行量化。在ICH恢复过程中,我们发现较晚出现了中度至重度血管痉挛,用尼莫地平进行了治疗。目前,患者临床状况正常。
HMG-CoA裂解酶缺乏症患者必须避免饮酒。在我们的患者中,苯乙酸钠和苯甲酸钠有效治疗了高氨血症。磁共振成像显示并量化了细胞毒性脑水肿。较晚出现的脑血管痉挛是脑损伤的另一种机制。这些观察结果此前均未被报道。