Department of Pediatrics, Yale University School of Medicine, New Haven, Conn.
Division of Immunology and Rheumatology, Department of Medicine, Stanford University School of Medicine, Stanford, Calif.
J Allergy Clin Immunol Pract. 2017 Sep-Oct;5(5):1344-1350.e3. doi: 10.1016/j.jaip.2017.01.028. Epub 2017 Mar 9.
Smith-Magenis syndrome (SMS) is a complex neurobehavioral disorder associated with recurrent otitis. Most SMS cases result from heterozygous interstitial chromosome 17p11.2 deletions that encompass not only the intellectual disability gene retinoic acid-induced 1 but also other genes associated with immunodeficiency, autoimmunity, and/or malignancy.
The goals of this study were to describe the immunological consequence of 17p11.2 deletions by determining the prevalence of immunological diseases in subjects with SMS and by assessing their immune systems via laboratory methods.
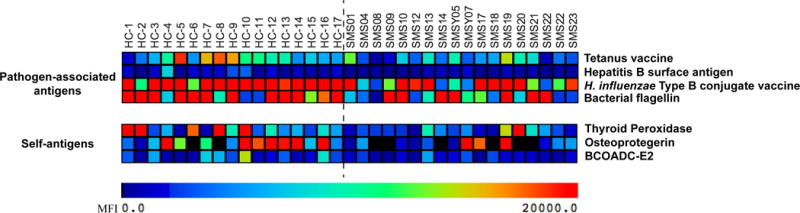
We assessed clinical histories of 76 subjects with SMS with heterozygous 17p11.2 deletions and performed in-depth immunological testing on 25 representative cohort members. Laboratory testing included determination of serum antibody concentrations, vaccine titers, and lymphocyte subset frequencies. Detailed reactivity profiles of SMS serum antibodies were performed using custom-made antigen microarrays.
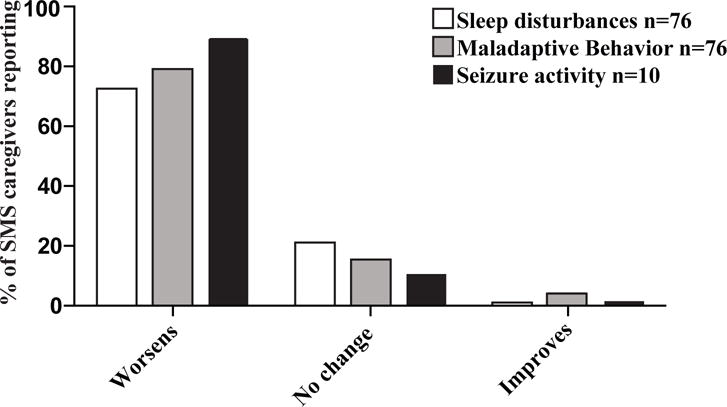
Of 76 subjects with SMS, 74 reported recurrent infections including otitis (88%), pneumonia (47%), sinusitis (42%), and gastroenteritis (34%). Infections were associated with worsening SMS-related neurobehavioral symptoms. The prevalence of autoimmune and atopic diseases was not increased. Malignancy was not reported. Laboratory evaluation revealed most subjects with SMS to be deficient of isotype-switched memory B cells and many to lack protective antipneumococcal antibodies. SMS antibodies were not more reactive than control antibodies to self-antigens.
Patients with SMS with heterozygous 17p.11.2 deletions display an increased susceptibility to sinopulmonary infections, but not to autoimmune, allergic, or malignant diseases. SMS sera display an antibody reactivity profile favoring neither recognition of pathogen-associated antigens nor self-antigens. Prophylactic strategies to prevent infections may also provide neurobehavioral benefits to selected patients with SMS.
Smith-Magenis 综合征(SMS)是一种与复发性中耳炎相关的复杂神经行为障碍。大多数 SMS 病例是由杂合性 17p11.2 染色体片段缺失引起的,这些缺失不仅包含智力障碍基因维甲酸诱导 1,还包含其他与免疫缺陷、自身免疫和/或恶性肿瘤相关的基因。
本研究旨在通过确定 SMS 患者免疫性疾病的患病率,并通过实验室方法评估其免疫系统,来描述 17p11.2 缺失的免疫学后果。
我们评估了 76 名携带 17p11.2 缺失杂合子的 SMS 患者的临床病史,并对 25 名代表性队列成员进行了深入的免疫学检测。实验室检测包括血清抗体浓度、疫苗效价和淋巴细胞亚群频率的测定。使用定制的抗原微阵列对 SMS 血清抗体的详细反应谱进行了分析。
在 76 名 SMS 患者中,74 名患者报告有复发性感染,包括中耳炎(88%)、肺炎(47%)、鼻窦炎(42%)和胃肠炎(34%)。感染与 SMS 相关神经行为症状的恶化有关。自身免疫性和特应性疾病的患病率没有增加。没有报告恶性肿瘤。实验室评估显示,大多数 SMS 患者存在免疫球蛋白转换记忆 B 细胞缺陷,许多患者缺乏保护性抗肺炎球菌抗体。SMS 抗体对自身抗原的反应性不比对照抗体更强烈。
携带 17p11.2 缺失杂合子的 SMS 患者易发生鼻窦肺部感染,但不易发生自身免疫、过敏或恶性疾病。SMS 血清显示出一种抗体反应谱,既不倾向于识别病原体相关抗原,也不倾向于识别自身抗原。预防感染的策略也可能为选定的 SMS 患者提供神经行为益处。