DeJesus Michael A, Nambi Subhalaxmi, Smith Clare M, Baker Richard E, Sassetti Christopher M, Ioerger Thomas R
Department of Computer Science, Texas A&M University, College Station, TX 77843, USA.
Department of Microbiology and Physiological Systems, University of Massachusetts Medical School, 55 Lake Avenue N., Worcester, MA 01655, USA.
Nucleic Acids Res. 2017 Jun 20;45(11):e93. doi: 10.1093/nar/gkx128.
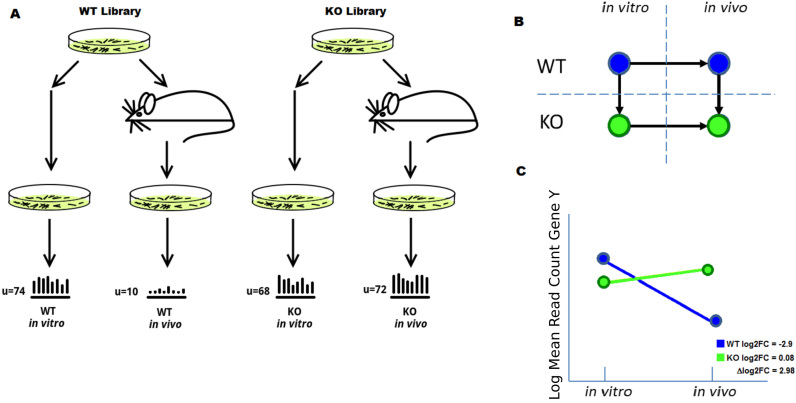
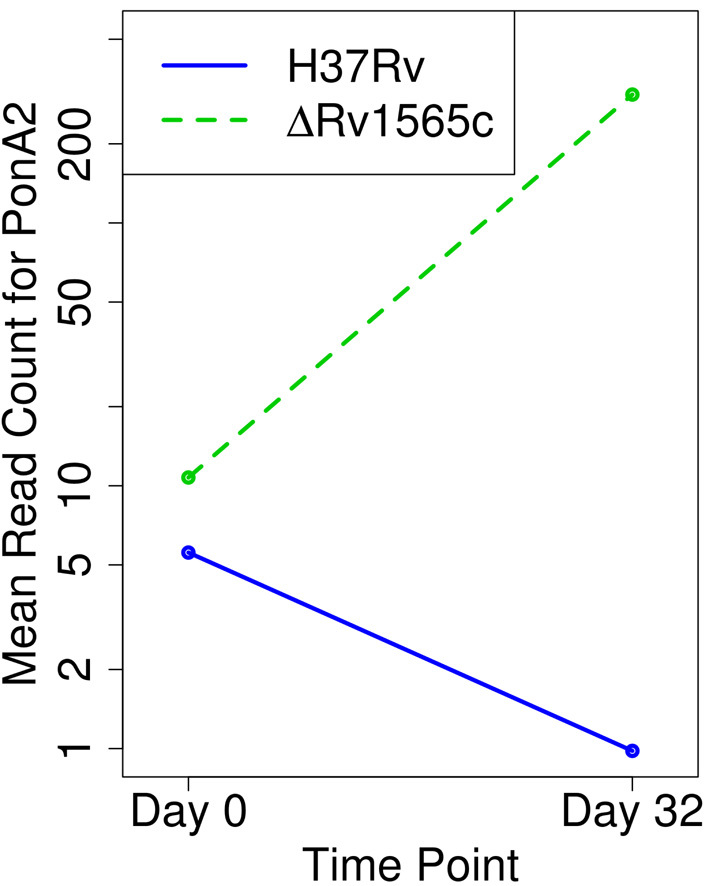
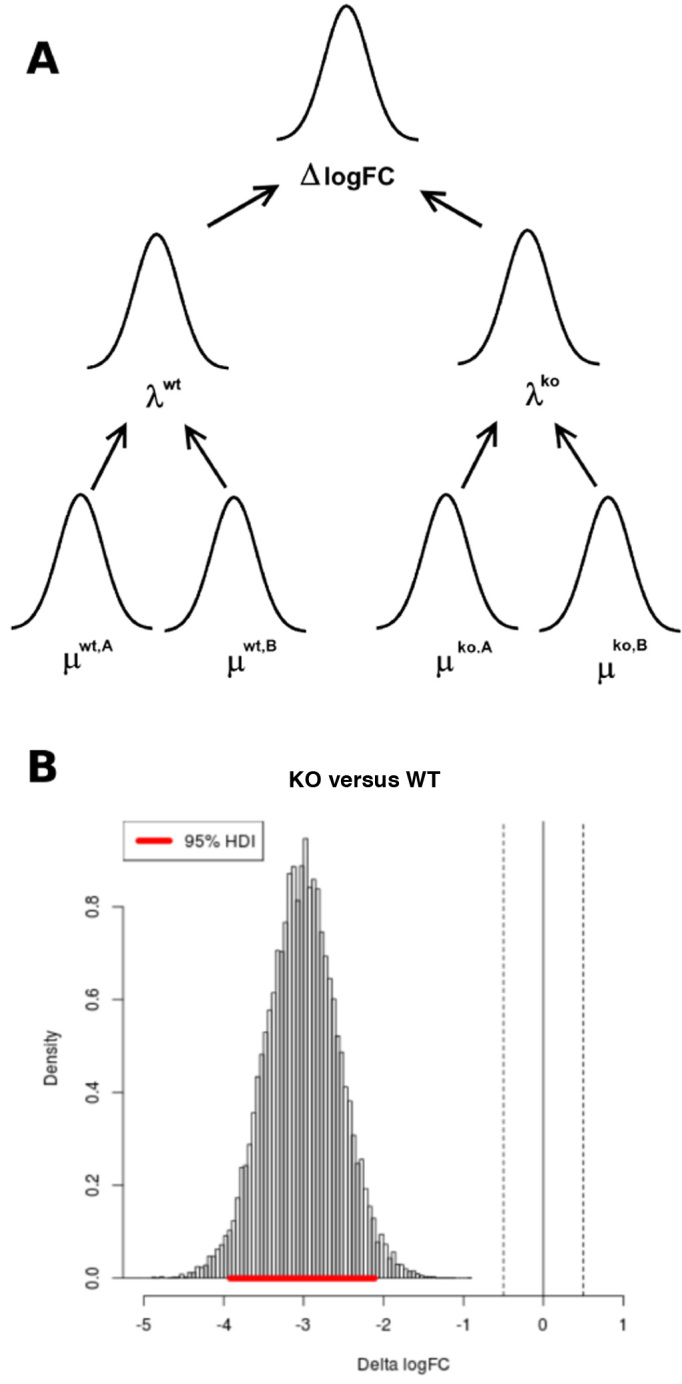
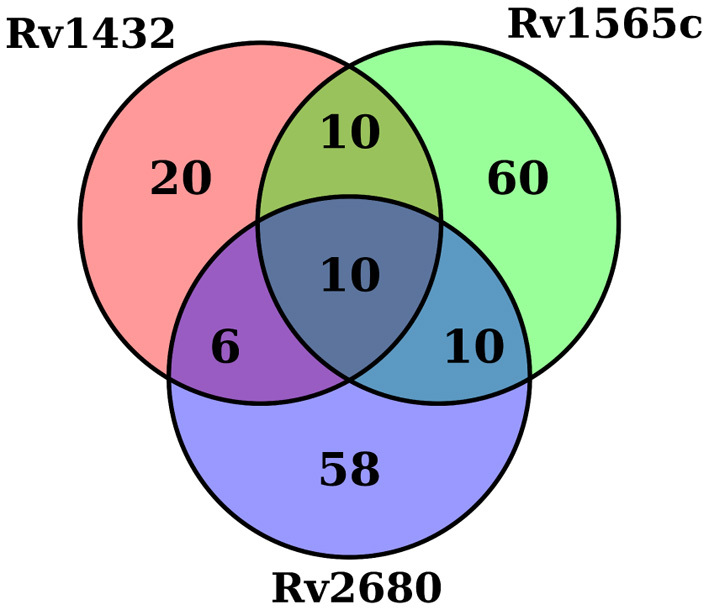
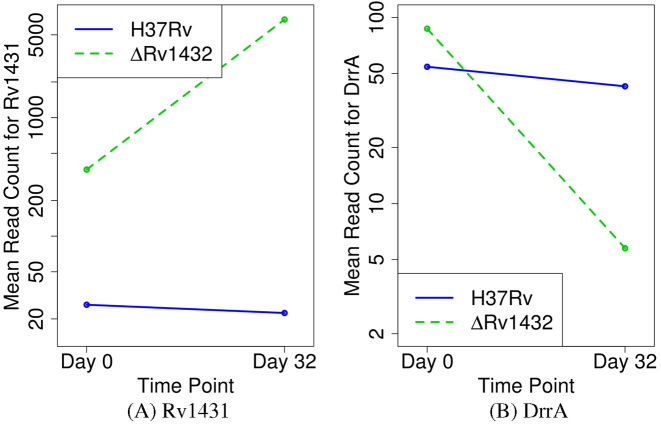
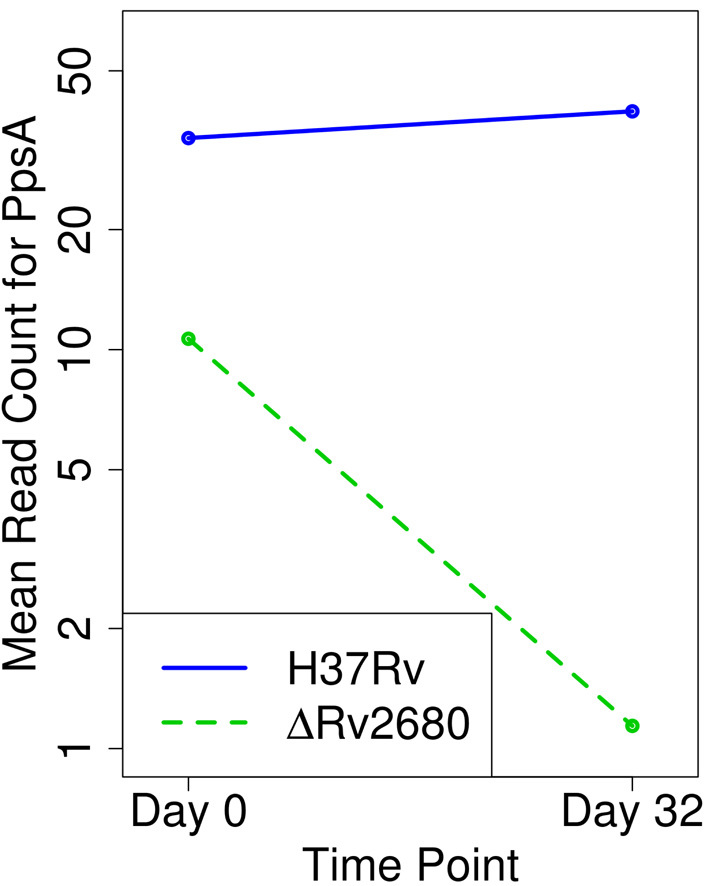
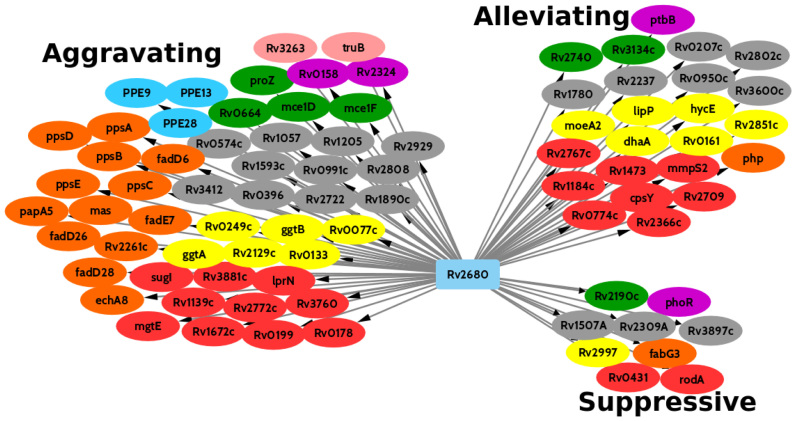
Tn-Seq is an experimental method for probing the functions of genes through construction of complex random transposon insertion libraries and quantification of each mutant's abundance using next-generation sequencing. An important emerging application of Tn-Seq is for identifying genetic interactions, which involves comparing Tn mutant libraries generated in different genetic backgrounds (e.g. wild-type strain versus knockout strain). Several analytical methods have been proposed for analyzing Tn-Seq data to identify genetic interactions, including estimating relative fitness ratios and fitting a generalized linear model. However, these have limitations which necessitate an improved approach. We present a hierarchical Bayesian method for identifying genetic interactions through quantifying the statistical significance of changes in enrichment. The analysis involves a four-way comparison of insertion counts across datasets to identify transposon mutants that differentially affect bacterial fitness depending on genetic background. Our approach was applied to Tn-Seq libraries made in isogenic strains of Mycobacterium tuberculosis lacking three different genes of unknown function previously shown to be necessary for optimal fitness during infection. By analyzing the libraries subjected to selection in mice, we were able to distinguish several distinct classes of genetic interactions for each target gene that shed light on their functions and roles during infection.
转座子测序(Tn-Seq)是一种实验方法,通过构建复杂的随机转座子插入文库,并使用下一代测序对每个突变体的丰度进行定量,来探究基因的功能。Tn-Seq一个重要的新兴应用是识别遗传相互作用,这涉及比较在不同遗传背景下(例如野生型菌株与基因敲除菌株)产生的Tn突变体文库。已经提出了几种分析方法来分析Tn-Seq数据以识别遗传相互作用,包括估计相对适合度比率和拟合广义线性模型。然而,这些方法存在局限性,需要一种改进的方法。我们提出了一种层次贝叶斯方法,通过量化富集变化的统计显著性来识别遗传相互作用。该分析涉及对各数据集的插入计数进行四路比较,以识别根据遗传背景不同程度影响细菌适合度的转座子突变体。我们的方法应用于在结核分枝杆菌同基因菌株中构建的Tn-Seq文库,这些菌株缺失三个功能未知的不同基因,先前已证明这些基因是感染期间最佳适合度所必需的。通过分析在小鼠体内经过选择的文库,我们能够区分每个靶基因的几类不同的遗传相互作用,从而揭示它们在感染过程中的功能和作用。