Nash William T, Gillespie Alyssa L, Brown Michael G
Department of Microbiology, Immunology, and Cancer Biology, School of Medicine, University of Virginia, Charlottesville, VA, USA; Beirne B. Carter Center for Immunology Research, School of Medicine, University of Virginia, Charlottesville, VA, USA; Division of Nephrology, Department of Medicine, University of Virginia, Charlottesville, VA, USA.
Beirne B. Carter Center for Immunology Research, School of Medicine, University of Virginia, Charlottesville, VA, USA; Division of Nephrology, Department of Medicine, University of Virginia, Charlottesville, VA, USA.
Front Immunol. 2017 Mar 9;8:251. doi: 10.3389/fimmu.2017.00251. eCollection 2017.
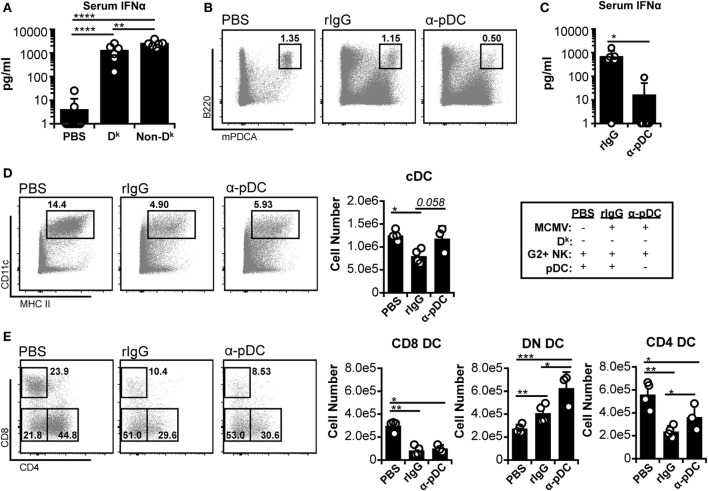
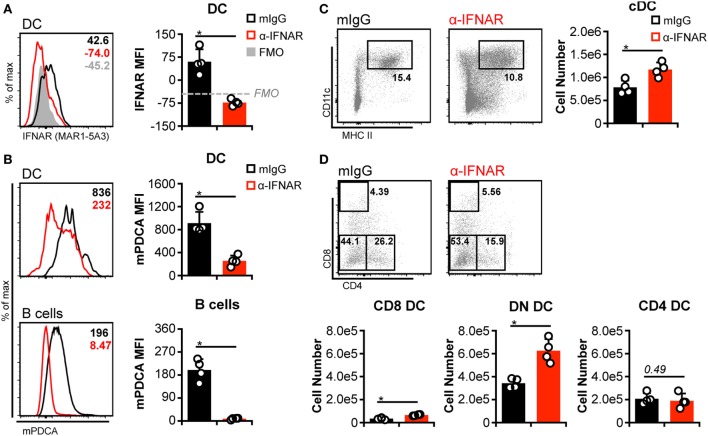
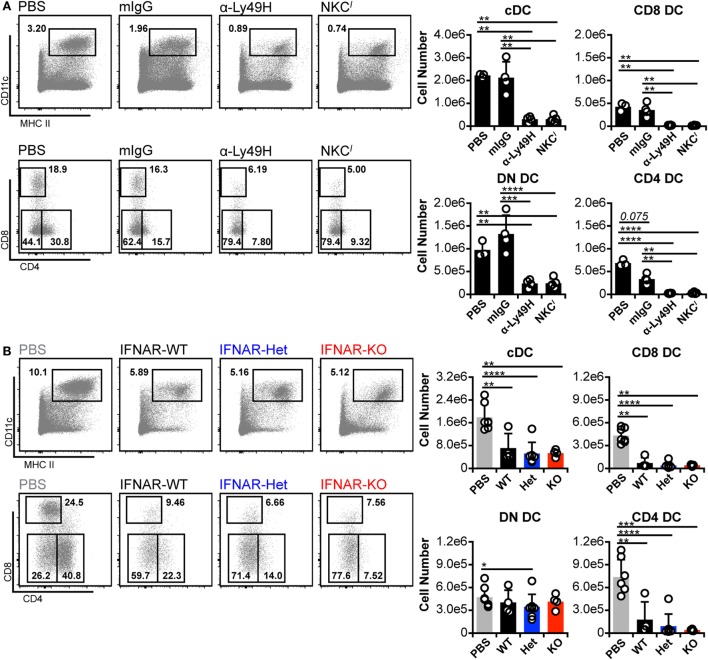
Dendritic cells (DC) are well-known modulators of immunity. This heterogeneous population is composed of defined subsets that exhibit functional specialization and are critical in initiating responses to pathogens. As such, many infectious agents employ strategies to disrupt DC functioning in attempts to evade the immune system. In some instances, this manifests as an outright loss of these cells. Previous work has suggested that, in the absence of an efficient natural killer (NK) cell response, murine cytomegalovirus (MCMV) induces large amounts of interferon (IFN)-I. This heightened IFN-I response is thought to contribute to conventional DC (cDC) loss and delayed development of T cell immunity. However, the precise role of IFN-I in such cDC loss remains unclear. We investigated the effects of licensed NK cells and IFN-I signaling on splenic cDC subsets during MCMV infection and found that a licensed NK cell response partially protects cDC numbers, but does not prevent increases in serum IFN-I. This suggested that high residual IFN-I could contribute to cDC loss. Therefore, we used multiple strategies to modulate IFN-I signaling during MCMV infection including plasmacytoid DC depletion, IFN-I receptor (IFNAR) blockade, and genetic ablation of IFNAR expression. Interestingly, restriction of IFN-I signals did not substantially preserve either CD8 or CD4 DC total numbers, but resulted in significant retention and/or accumulation of the splenic CD8 CD4 [double negative (DN)] subset. However, the DN DC effect manifested in a DC-extrinsic manner since IFNAR-deficient cells were not preferentially retained over their IFNAR wild-type counterparts in a mixed-chimera setting. Our results show that IFN-I signaling is not responsible for overt cDC toxicity in the setting of acute MCMV infection and emphasize that additional mechanisms contribute to DC loss and require exploration.
树突状细胞(DC)是众所周知的免疫调节因子。这个异质性群体由特定的亚群组成,这些亚群表现出功能特化,并且在启动对病原体的反应中至关重要。因此,许多感染因子采用策略来破坏DC的功能,试图逃避免疫系统。在某些情况下,这表现为这些细胞的完全丧失。先前的研究表明,在缺乏有效的自然杀伤(NK)细胞反应的情况下,小鼠巨细胞病毒(MCMV)会诱导大量的I型干扰素(IFN)。这种增强的IFN-I反应被认为有助于传统DC(cDC)的丧失和T细胞免疫的延迟发展。然而,IFN-I在这种cDC丧失中的精确作用仍不清楚。我们研究了许可的NK细胞和IFN-I信号在MCMV感染期间对脾脏cDC亚群的影响,发现许可的NK细胞反应部分保护cDC数量,但不能阻止血清IFN-I的增加。这表明高残留的IFN-I可能导致cDC丧失。因此,我们在MCMV感染期间使用了多种策略来调节IFN-I信号,包括浆细胞样DC耗竭、IFN-I受体(IFNAR)阻断和IFNAR表达的基因消融。有趣的是,限制IFN-I信号并没有实质性地保留CD8或CD4 DC的总数,但导致脾脏CD8 CD4 [双阴性(DN)]亚群的显著保留和/或积累。然而,DN DC效应以DC外在的方式表现出来,因为在混合嵌合体环境中,IFNAR缺陷细胞并不比其IFNAR野生型对应物更优先地被保留。我们的结果表明,在急性MCMV感染的情况下,IFN-I信号并不负责明显的cDC毒性,并强调其他机制导致DC丧失,需要进一步探索。