Martin Simon H, Van Belleghem Steven M
Department of Zoology, University of Cambridge, CB2 3EJ, United Kingdom
Department of Zoology, University of Cambridge, CB2 3EJ, United Kingdom.
Genetics. 2017 May;206(1):429-438. doi: 10.1534/genetics.116.194720. Epub 2017 Mar 24.
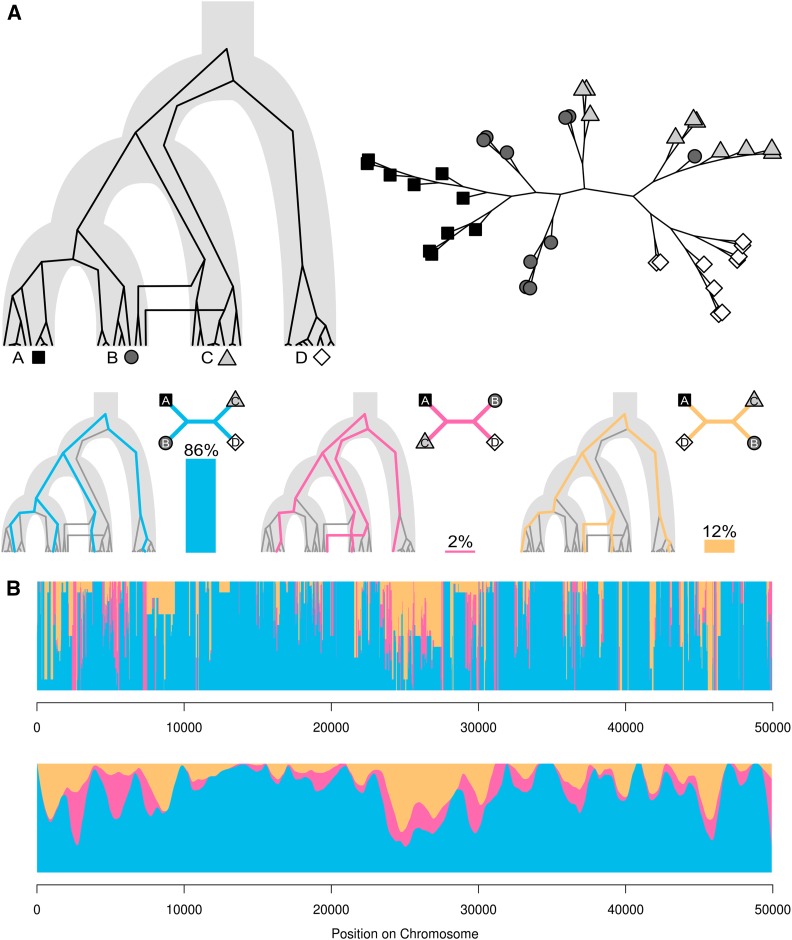
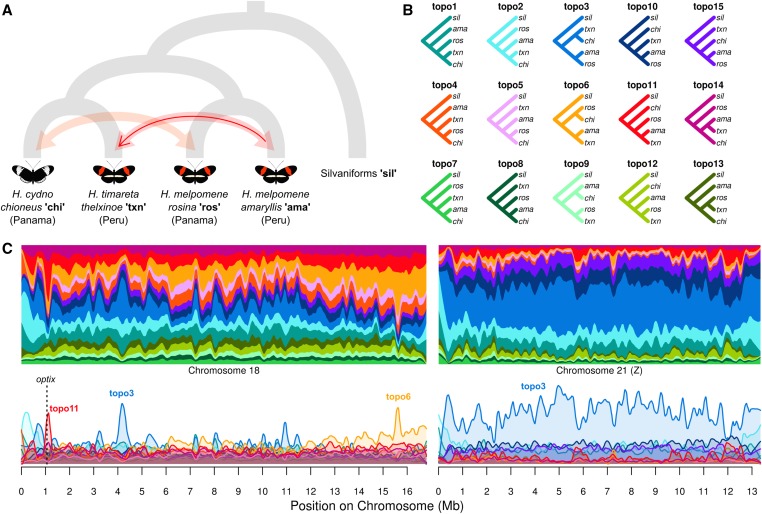
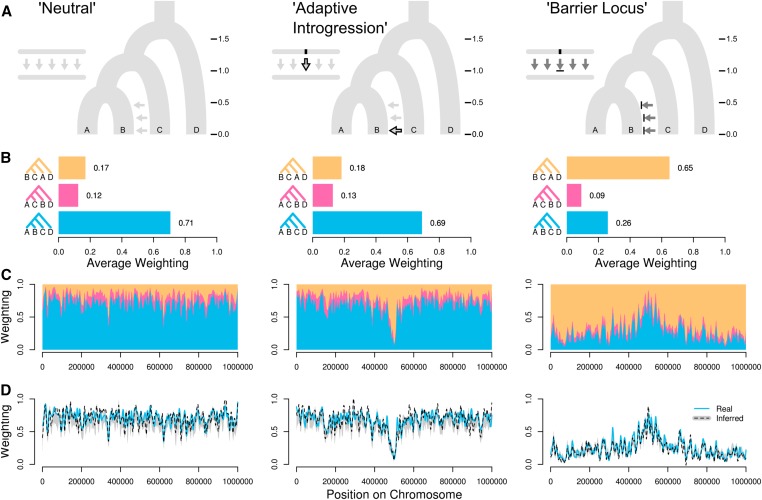
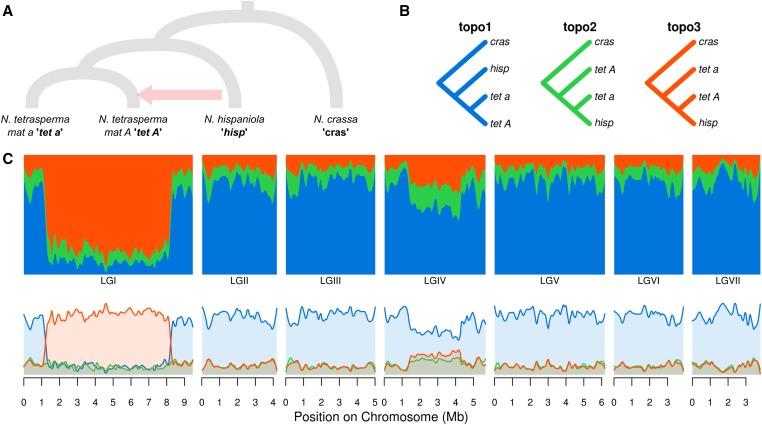
We introduce the concept of topology weighting, a method for quantifying relationships between taxa that are not necessarily monophyletic, and visualizing how these relationships change across the genome. A given set of taxa can be related in a limited number of ways, but if each taxon is represented by multiple sequences, the number of possible topologies becomes very large. Topology weighting reduces this complexity by quantifying the contribution of each taxon topology to the full tree. We describe our method for topology weighting by iterative sampling of subtrees (), and test it on both simulated and real genomic data. Overall, we show that this is an informative and versatile approach, suitable for exploring relationships in almost any genomic dataset. Scripts to implement the method described are available at http://github.com/simonhmartin/twisst.
我们引入了拓扑加权的概念,这是一种用于量化不一定是单系类群之间关系的方法,并可视化这些关系如何在整个基因组中变化。给定的一组类群可以通过有限的方式相互关联,但如果每个类群由多个序列表示,那么可能的拓扑结构数量就会变得非常大。拓扑加权通过量化每个类群拓扑对完整树的贡献来降低这种复杂性。我们通过对 subtree() 进行迭代采样来描述我们的拓扑加权方法,并在模拟和真实基因组数据上对其进行测试。总体而言,我们表明这是一种信息丰富且通用的方法,适用于探索几乎任何基因组数据集中的关系。实现所述方法的脚本可在 http://github.com/simonhmartin/twisst 获得。