McCulloch Derek, Brown Christina, Iland Harry
Institute of Hematology, Royal Prince Alfred Hospital, Camperdown, NSW, Australia.
Onco Targets Ther. 2017 Mar 14;10:1585-1601. doi: 10.2147/OTT.S100513. eCollection 2017.
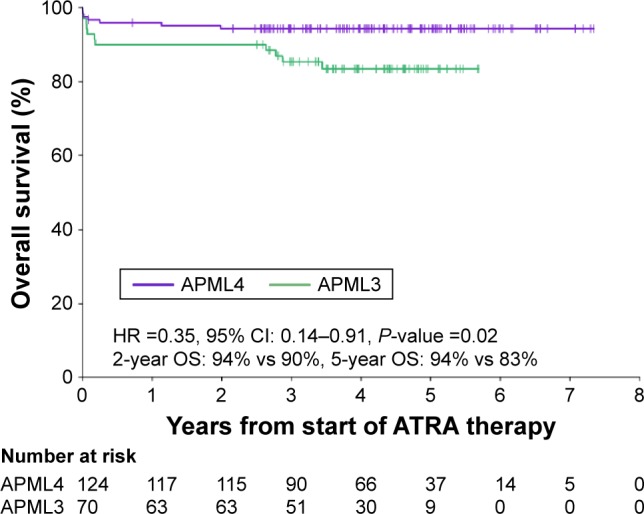
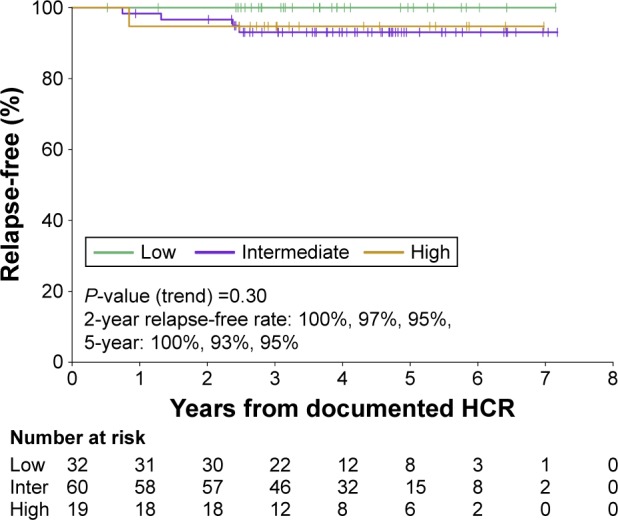
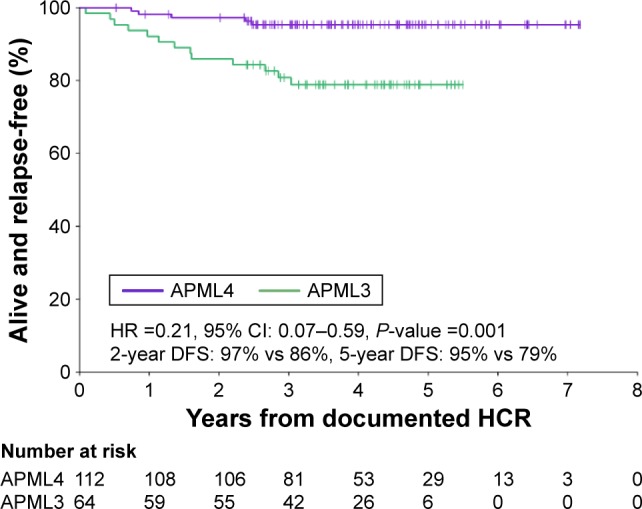
Acute promyelocytic leukemia (APL) is a distinct subtype of acute myeloid leukemia (AML) with a unique morphological appearance, associated coagulopathy and canonical balanced translocation of genetic material between chromosomes 15 and 17. APL was first described as a distinct subtype of AML in 1957 by Dr Leif Hillestad who recognized the pattern of an acute leukemia associated with fibrinolysis, hypofibrinogenemia and catastrophic hemorrhage. In the intervening years, the characteristic morphology of APL has been described fully with both classical hypergranular and variant microgranular forms. Both are characterized by a balanced translocation between the long arms of chromosomes 15 and 17, [t(15;17)(q24;q21)], giving rise to a unique fusion gene and an abnormal chimeric transcription factor (PML-RARA), which disrupts normal myeloid differentiation programs. The success of current treatments for APL is in marked contrast to the vast majority of patients with non-promyelocytic AML. The overall prognosis in non-promyelocytic AML is poor, and although there has been an improvement in overall survival in patients aged <60 years, only 30%-40% of younger patients are still alive 5 years after diagnosis. APL therapy has diverged from standard AML therapy through the empirical discovery of two agents that directly target the molecular basis of the disease. The evolution of treatment over the last 4 decades to include all- retinoic acid and arsenic trioxide, with chemotherapy limited to patients with high-risk disease, has led to complete remission in 90%-100% of patients in trials and rates of overall survival between 86% and 97%.
急性早幼粒细胞白血病(APL)是急性髓系白血病(AML)的一种独特亚型,具有独特的形态学表现、相关的凝血病以及15号和17号染色体之间遗传物质的典型平衡易位。1957年,Leif Hillestad博士首次将APL描述为AML的一种独特亚型,他认识到一种与纤维蛋白溶解、低纤维蛋白原血症和灾难性出血相关的急性白血病模式。在随后的几年里,APL的特征性形态已被充分描述,包括经典的高颗粒型和变异的微颗粒型。两者的特征都是15号和17号染色体长臂之间的平衡易位,[t(15;17)(q24;q21)],产生一个独特的融合基因和一个异常的嵌合转录因子(PML-RARA),它破坏了正常的髓系分化程序。目前APL治疗的成功与绝大多数非早幼粒细胞AML患者形成鲜明对比。非早幼粒细胞AML的总体预后很差,尽管<60岁患者的总生存率有所提高,但只有30%-40%的年轻患者在诊断后5年仍存活。APL治疗已与标准AML治疗不同,通过经验性发现两种直接针对该疾病分子基础的药物。在过去40年中,治疗方法不断演变,包括全反式维甲酸和三氧化二砷,化疗仅限于高危疾病患者,这使得试验中90%-100%的患者完全缓解,总生存率在86%至97%之间。