Phan Lon, Hsu Jeffrey, Tri Le Quang Minh, Willi Michaela, Mansour Tamer, Kai Yan, Garner John, Lopez John, Busby Ben
National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA.
Cleveland Clinic Lerner Research Institute, Cleveland, OH, USA.
F1000Res. 2016 Apr 13;5:673. doi: 10.12688/f1000research.8290.2. eCollection 2016.
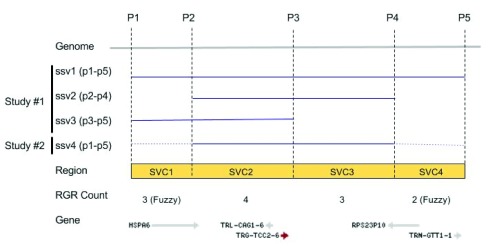
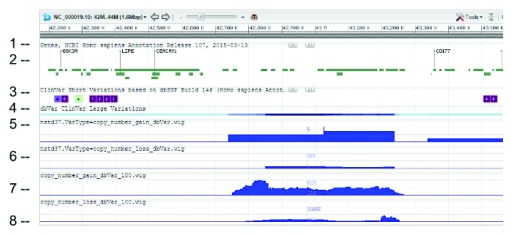
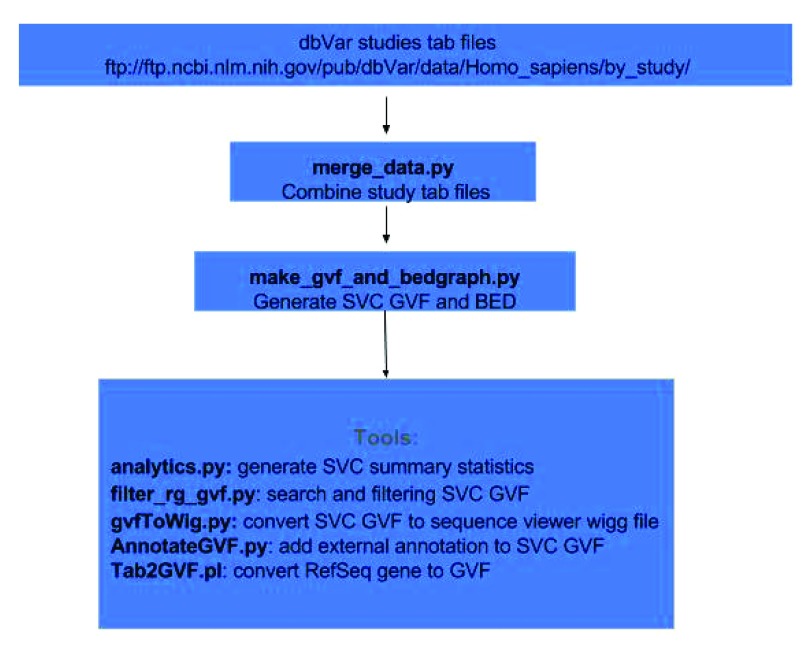
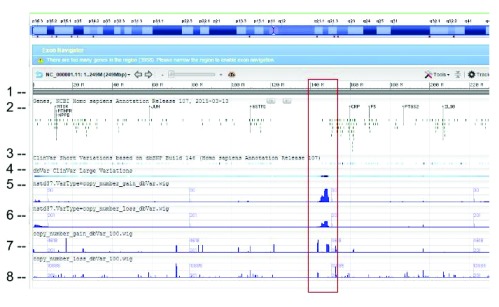
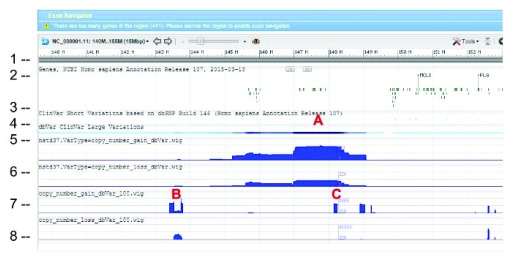
dbVar houses over 3 million submitted structural variants (SSV) from 120 human studies including copy number variations (CNV), insertions, deletions, inversions, translocations, and complex chromosomal rearrangements. Users can submit multiple SSVs to dbVAR that are presumably identical, but were ascertained by different platforms and samples, to calculate whether the variant is rare or common in the population and allow for cross validation. However, because SSV genomic location reporting can vary - including fuzzy locations where the start and/or end points are not precisely known - analysis, comparison, annotation, and reporting of SSVs across studies can be difficult. This project was initiated by the Structural Variant Comparison Group for the purpose of generating a non-redundant set of genomic regions defined by counts of concordance for all human SSVs placed on RefSeq assembly GRCh38 (RefSeq accession GCF_000001405.26). We intend that the availability of these regions, called structural variant clusters (SVCs), will facilitate the analysis, annotation, and exchange of SV data and allow for simplified display in genomic sequence viewers for improved variant interpretation. Sets of SVCs were generated by variant type for each of the 120 studies as well as for a combined set across all studies. Starting from 3.64 million SSVs, 2.5 million and 3.4 million non-redundant SVCs with count >=1 were generated by variant type for each study and across all studies, respectively. In addition, we have developed utilities for annotating, searching, and filtering SVC data in GVF format for computing summary statistics, exporting data for genomic viewers, and annotating the SVC using external data sources.
dbVar收录了来自120项人类研究的300多万个已提交的结构变异(SSV),包括拷贝数变异(CNV)、插入、缺失、倒位、易位和复杂染色体重排。用户可以向dbVAR提交多个可能相同但由不同平台和样本确定的SSV,以计算该变异在人群中是罕见还是常见,并进行交叉验证。然而,由于SSV基因组位置报告可能存在差异——包括起始和/或终点不精确已知的模糊位置——跨研究的SSV分析、比较、注释和报告可能会很困难。该项目由结构变异比较小组发起,目的是生成一组非冗余的基因组区域,这些区域由放置在RefSeq组装GRCh38(RefSeq登录号GCF_000001405.26)上的所有人类SSV的一致性计数定义。我们希望这些称为结构变异簇(SVC)的区域的可用性将促进SV数据的分析、注释和交换,并允许在基因组序列查看器中进行简化显示,以改进变异解释。针对120项研究中的每一项以及所有研究的组合集,按变异类型生成了SVC集。从364万个SSV开始,每项研究和所有研究按变异类型分别生成了250万个和340万个计数≥1的非冗余SVC。此外,我们还开发了实用工具,用于以GVF格式注释、搜索和过滤SVC数据,以计算汇总统计信息、导出基因组查看器的数据以及使用外部数据源注释SVC。