Wang Rui, Deng Xiaolan, Yoshioka Yuichiro, Vougiouklakis Theodore, Park Jae-Hyun, Suzuki Takehiro, Dohmae Naoshi, Ueda Koji, Hamamoto Ryuji, Nakamura Yusuke
Section of Hematology/Oncology, Department of Medicine, The University of Chicago, Chicago, Illinois, USA.
State Key Laboratory of Cancer Biology and Xijing Hospital of Digestive Diseases, Xijing Hospital, Fourth Military Medical University, Xi'an, China.
Cancer Sci. 2017 Jun;108(6):1203-1209. doi: 10.1111/cas.13245.
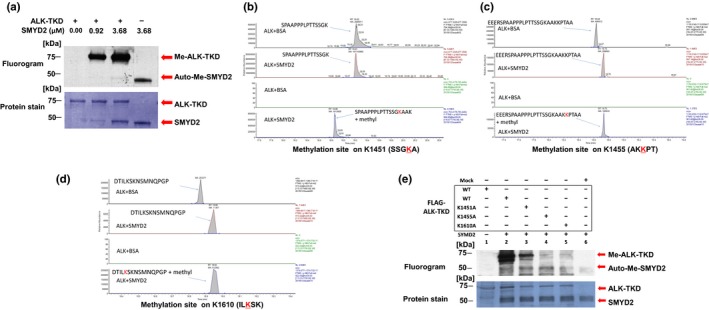
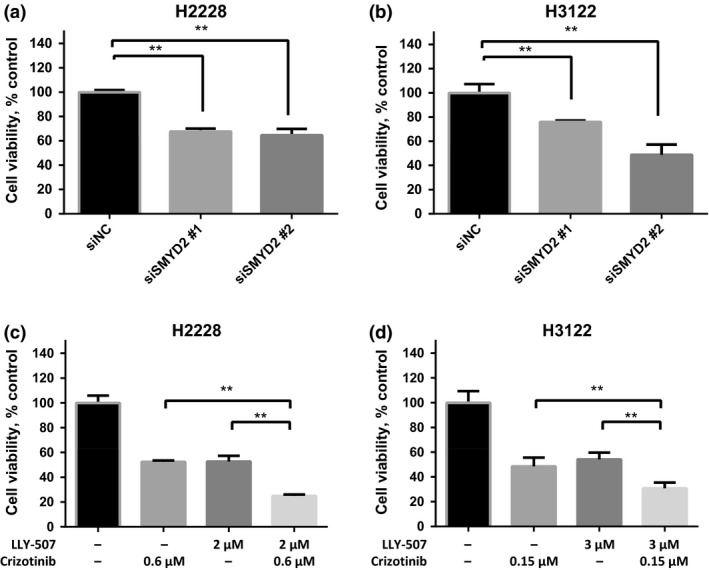
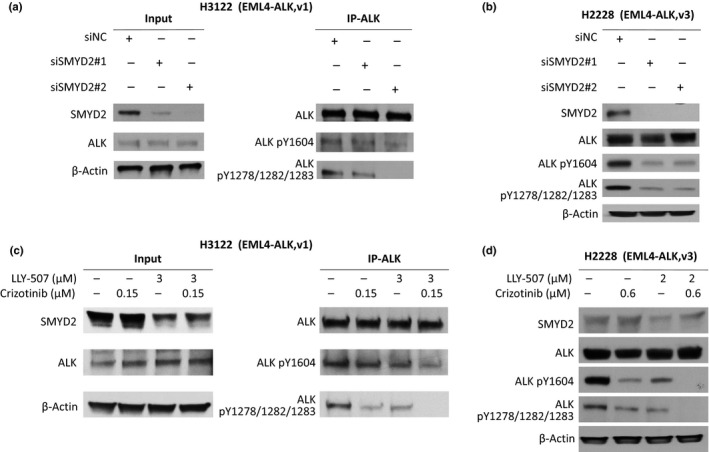
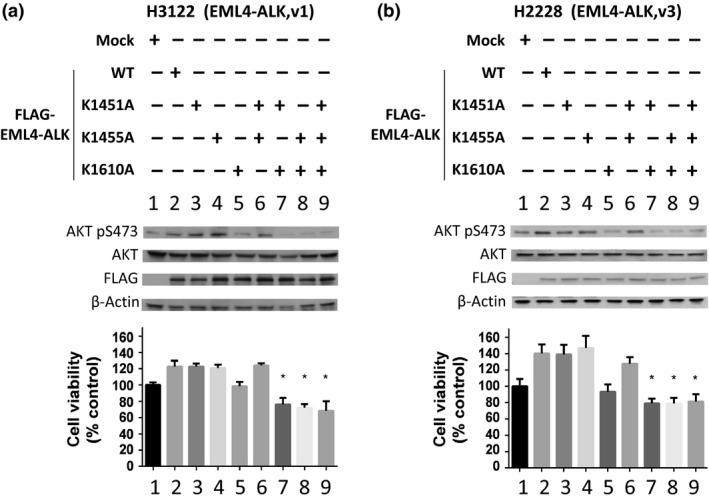
A specific subtype of non-small-cell lung cancer (NSCLC) characterized with an EML4-ALK fusion gene, which drives constitutive oncogenic activation of anaplastic lymphoma kinase (ALK), shows a good clinical response to ALK inhibitors. We have reported multiple examples implying the biological significance of methylation on non-histone proteins including oncogenic kinases in human carcinogenesis. Through the process to search substrates for various methyltransferases using an in vitro methyltransferase assay, we found that a lysine methyltransferase, SET and MYND domain-containing 2 (SMYD2), could methylate lysine residues 1451, 1455, and 1610 in ALK protein. Knockdown of SMYD2 as well as treatment with a SMYD2 inhibitor in two NSCLC cell lines with an EML4-ALK gene significantly attenuated the phosphorylation levels of the EML4-ALK protein. Substitutions of each of these three lysine residues to an alanine partially or almost completely diminished in vitro methylation of ALK. In addition, we found that exogenous introduction of EML4-ALK protein with the substitution of lysine 1610 to an alanine in these two cell lines reduced the phosphorylation levels of AKT, one of the downstream oncogenic molecules in the EML4-ALK pathway, and suppressed the growth of the two cell lines. We further showed that the combination of a SMYD2 inhibitor and an ALK inhibitor additively suppressed the growth of these two NSCLC cells, compared with single-agent treatment. Our results shed light on a novel mechanism that modulates the kinase activity of the ALK fused gene product and imply that SMYD2-mediated ALK methylation might be a promising target for development of a novel class of treatment for tumors with the ALK fused gene.
一种以EML4-ALK融合基因为特征的非小细胞肺癌(NSCLC)亚型,该融合基因驱动间变性淋巴瘤激酶(ALK)的组成性致癌激活,对ALK抑制剂显示出良好的临床反应。我们已经报道了多个例子,暗示了甲基化在人类致癌过程中对包括致癌激酶在内的非组蛋白的生物学意义。通过使用体外甲基转移酶测定法寻找各种甲基转移酶底物的过程,我们发现一种赖氨酸甲基转移酶,含SET和MYND结构域2(SMYD2),可以使ALK蛋白中的赖氨酸残基1451、1455和1610甲基化。在两个具有EML4-ALK基因的NSCLC细胞系中敲低SMYD2以及用SMYD2抑制剂处理显著降低了EML4-ALK蛋白的磷酸化水平。将这三个赖氨酸残基中的每一个替换为丙氨酸可部分或几乎完全减少ALK的体外甲基化。此外,我们发现在这两个细胞系中外源引入将赖氨酸1610替换为丙氨酸的EML4-ALK蛋白降低了AKT的磷酸化水平,AKT是EML4-ALK途径中的下游致癌分子之一,并抑制了这两个细胞系的生长。我们进一步表明,与单药治疗相比,SMYD2抑制剂和ALK抑制剂联合使用可加成抑制这两种NSCLC细胞的生长。我们的结果揭示了一种调节ALK融合基因产物激酶活性的新机制,并暗示SMYD2介导的ALK甲基化可能是开发针对具有ALK融合基因的肿瘤的新型治疗方法的有希望的靶点。