Aguisanda Francis, Thorne Natasha, Zheng Wei
National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, MD 20892-3370, USA.
Curr Chem Genom Transl Med. 2017 Jan 30;11:1-18. doi: 10.2174/2213988501711010001. eCollection 2017.
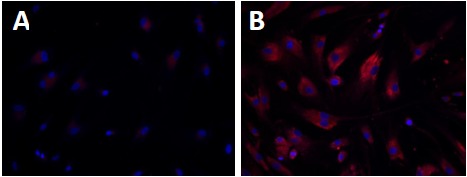
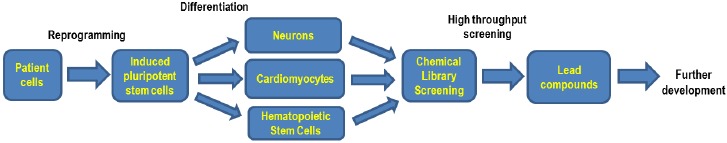
Wolman disease (WD) and cholesteryl ester storage disease (CESD) are lysosomal storage diseases (LSDs) caused by a deficiency in lysosomal acid lipase (LAL) due to mutations in the gene. This enzyme is critical to the proper degradation of cholesterol in the lysosome. LAL function is completely lost in WD while some residual activity remains in CESD. Both are rare diseases with an incidence rate of less than 1/100,000 births for WD and approximate 2.5/100,000 births for CESD. Clinical manifestation of WD includes hepatosplenomegaly, calcified adrenal glands, severe malabsorption and a failure to thrive. As in CESD, histological analysis of WD tissues reveals the accumulation of triglycerides (TGs) and esterified cholesterol (EC) in cellular lysosomes. However, the clinical presentation of CESD is less severe and more variable than WD. This review is to provide an overview of the disease pathophysiology and the current state of therapeutic development for both of WD and CESD. The review will also discuss the application of patient derived iPSCs for further drug discovery.
沃尔曼病(WD)和胆固醇酯贮积病(CESD)是溶酶体贮积症(LSDs),由基因突变导致溶酶体酸性脂肪酶(LAL)缺乏引起。这种酶对溶酶体中胆固醇的正常降解至关重要。在WD中LAL功能完全丧失,而在CESD中仍保留一些残余活性。两者均为罕见病,WD的发病率低于1/100,000活产,CESD的发病率约为2.5/100,000活产。WD的临床表现包括肝脾肿大、肾上腺钙化、严重吸收不良和发育不良。与CESD一样,WD组织的组织学分析显示细胞溶酶体中甘油三酯(TGs)和酯化胆固醇(EC)的积累。然而,CESD的临床表现比WD轻且更具变异性。本综述旨在概述WD和CESD的疾病病理生理学以及治疗进展的现状。该综述还将讨论患者来源的诱导多能干细胞在进一步药物发现中的应用。