de Las Heras Javier, Almohalla Carolina, Blasco-Alonso Javier, Bourbon Mafalda, Couce Maria-Luz, de Castro López María José, García Jiménez Mª Concepción, Gil Ortega David, González-Diéguez Luisa, Meavilla Silvia, Moreno-Álvarez Ana, Pastor-Rosado José, Sánchez-Pintos Paula, Serrano-Gonzalo Irene, López Eduardo, Valdivielso Pedro, Yahyaoui Raquel, Quintero Jesús
Division of Pediatric Metabolism, Cruces University Hospital, CIBER-ER, Metab-ERN, University of the Basque Country (UPV/EHU), Biobizkaia Health Research Institute, 48903 Bilbao, Spain.
Unidad de Hepatología, Hospital Universitario Río Hortega, 47012 Valladolid, Spain.
Nutrients. 2024 Dec 13;16(24):4309. doi: 10.3390/nu16244309.
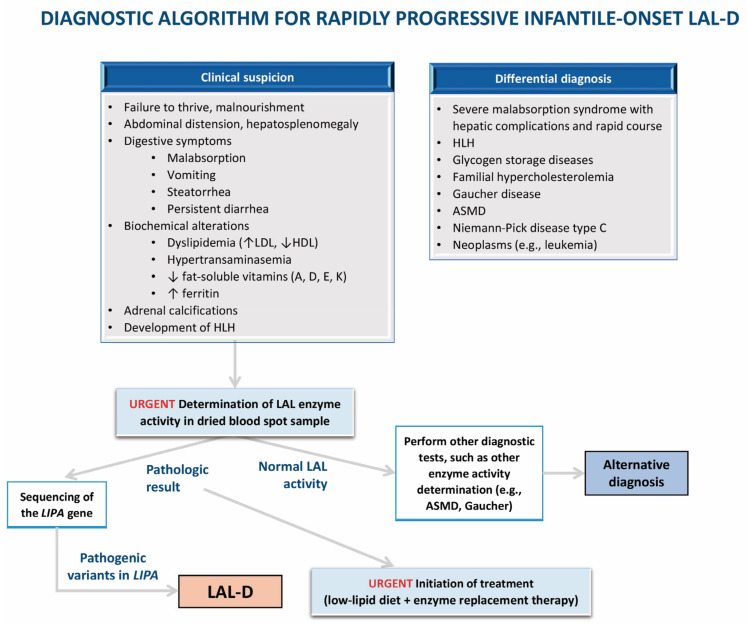
Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease with two distinct phenotypes, an infantile-onset form (formerly Wolman disease) and a later-onset form (formerly cholesteryl ester storage disease). The objective of this narrative review is to examine the most important aspects of the diagnosis and treatment of LAL-D and to provide practical expert recommendations. The infantile-onset form occurs in the first weeks of life and is characterized by malnourishment and failure to thrive due to gastrointestinal impairment (vomiting, diarrhea, malabsorption), as well as systemic inflammation, hepatosplenomegaly, and adrenal calcifications. Mortality is close to 100% before one year of life in the absence of specific treatment. The later-onset form can be diagnosed in childhood or adulthood and is characterized by chronic liver injury and/or lipid profile alterations. When LAL-D is suspected, enzyme activity should be determined to confirm the diagnosis, with analysis from a dried blood spot sample being the quickest and most reliable method. In infantile-onset LAL-D, the initiation of enzyme replacement therapy (sebelipase α) and careful nutritional management with a low-lipid diet is very urgent, as prognosis is directly linked to the early initiation of specific treatment. In recent years, our knowledge of the management of LAL-D has increased considerably, with improvements regarding the initial enzyme replacement therapy dose and careful nutritional treatment with a low-lipid diet to decrease lipid deposition and systemic inflammation, leading to better outcomes. In this narrative review we offer a quick guide for the initial management of infantile-onset LAL-D.
溶酶体酸性脂肪酶缺乏症(LAL-D)是一种极为罕见的溶酶体贮积病,有两种不同的表型,即婴儿型(以前称为沃尔曼病)和晚发型(以前称为胆固醇酯贮积病)。本叙述性综述的目的是探讨LAL-D诊断和治疗的最重要方面,并提供实用的专家建议。婴儿型在出生后的头几周出现,其特征是由于胃肠道功能受损(呕吐、腹泻、吸收不良)导致营养不良和发育不良,以及全身炎症、肝脾肿大和肾上腺钙化。在没有特异性治疗的情况下,一岁前的死亡率接近100%。晚发型可在儿童期或成人期诊断,其特征是慢性肝损伤和/或血脂改变。当怀疑患有LAL-D时,应测定酶活性以确诊,干血斑样本分析是最快且最可靠的方法。在婴儿型LAL-D中,启动酶替代疗法(sebelipase α)并采用低脂饮食进行仔细的营养管理非常紧迫,因为预后与特异性治疗的早期启动直接相关。近年来,我们对LAL-D管理的认识有了显著提高,在初始酶替代疗法剂量以及采用低脂饮食进行仔细的营养治疗以减少脂质沉积和全身炎症方面都有改进,从而带来了更好的结果。在本叙述性综述中,我们为婴儿型LAL-D的初始管理提供了快速指南。