Afyounian Ebrahim, Annala Matti, Nykter Matti
Faculty of Medicine and Life Sciences and BioMediTech institute, University of Tampere, Tampere, Finland.
BMC Bioinformatics. 2017 Apr 13;18(1):215. doi: 10.1186/s12859-017-1626-8.
Somatic alterations, including loss of heterozygosity, can affect the expression of oncogenes and tumor suppressor genes. Whole genome sequencing enables detailed characterization of such aberrations. However, due to the limitations of current high throughput sequencing technologies, this task remains challenging. Hence, accurate and reliable detection of such events is crucial for the identification of cancer-related alterations.
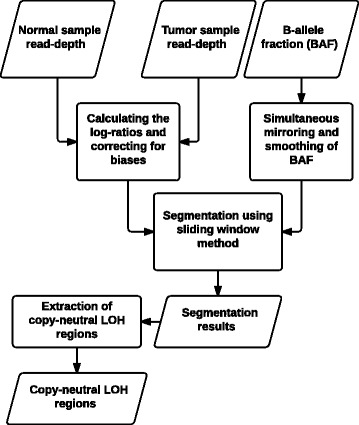
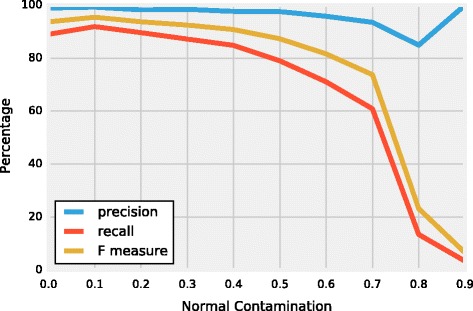
We introduce a new tool called Segmentum for determining somatic copy numbers using whole genome sequencing from paired tumor/normal samples. In our approach, read depth and B-allele fraction signals are smoothed, and double sliding windows are used to detect breakpoints, which makes our approach fast and straightforward. Because the breakpoint detection is performed simultaneously at different scales, it allows accurate detection as suggested by the evaluation results from simulated and real data. We applied Segmentum to paired tumor/normal whole genome sequencing samples from 38 patients with low-grade glioma from the TCGA dataset and were able to confirm the recurrence of copy-neutral loss of heterozygosity in chromosome 17p in low-grade astrocytoma characterized by IDH1/2 mutation and lack of 1p/19q co-deletion, which was previously reported using SNP array data.
Segmentum is an accurate, user-friendly tool for somatic copy number analysis of tumor samples. We demonstrate that this tool is suitable for the analysis of large cohorts, such as the TCGA dataset.
体细胞改变,包括杂合性缺失,可影响癌基因和肿瘤抑制基因的表达。全基因组测序能够详细表征此类畸变。然而,由于当前高通量测序技术的局限性,这项任务仍然具有挑战性。因此,准确可靠地检测此类事件对于识别癌症相关改变至关重要。
我们引入了一种名为Segmentum的新工具,用于使用配对肿瘤/正常样本的全基因组测序来确定体细胞拷贝数。在我们的方法中,读取深度和B等位基因分数信号被平滑处理,并使用双滑动窗口来检测断点,这使得我们的方法快速且直接。由于断点检测是在不同尺度上同时进行的,根据模拟和真实数据的评估结果,它能够进行准确检测。我们将Segmentum应用于来自TCGA数据集的38例低级别胶质瘤患者的配对肿瘤/正常全基因组测序样本,并能够证实先前使用SNP阵列数据报道的、以IDH1/2突变和缺乏1p/19q共缺失为特征的低级别星形细胞瘤中17号染色体短臂上拷贝数中性杂合性缺失的复发情况。
Segmentum是一种用于肿瘤样本体细胞拷贝数分析的准确、用户友好的工具。我们证明该工具适用于分析大型队列,如TCGA数据集。