Yu Zhenhua, Liu Yuanning, Shen Yi, Wang Minghui, Li Ao
School of Information Science and Technology and Centers for Biomedical Engineering, University of Science and Technology of China, Hefei AH230027, China.
School of Information Science and Technology and Centers for Biomedical Engineering, University of Science and Technology of China, Hefei AH230027, China School of Information Science and Technology and Centers for Biomedical Engineering, University of Science and Technology of China, Hefei AH230027, China.
Bioinformatics. 2014 Sep 15;30(18):2576-83. doi: 10.1093/bioinformatics/btu346. Epub 2014 May 19.
Whole-genome sequencing of tumor samples has been demonstrated as an efficient approach for comprehensive analysis of genomic aberrations in cancer genome. Critical issues such as tumor impurity and aneuploidy, GC-content and mappability bias have been reported to complicate identification of copy number alteration and loss of heterozygosity in complex tumor samples. Therefore, efficient computational methods are required to address these issues.
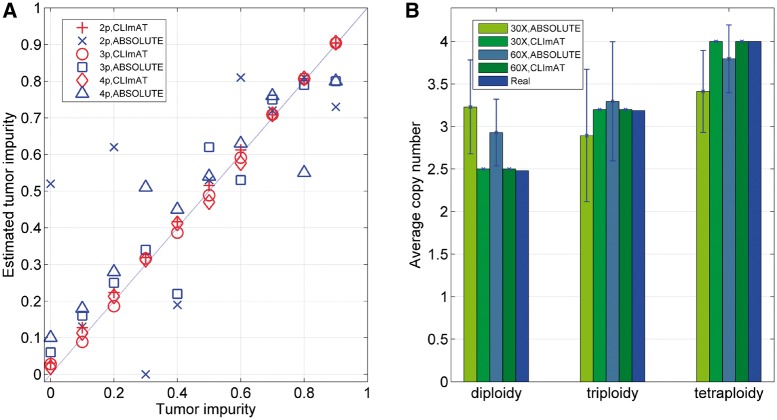
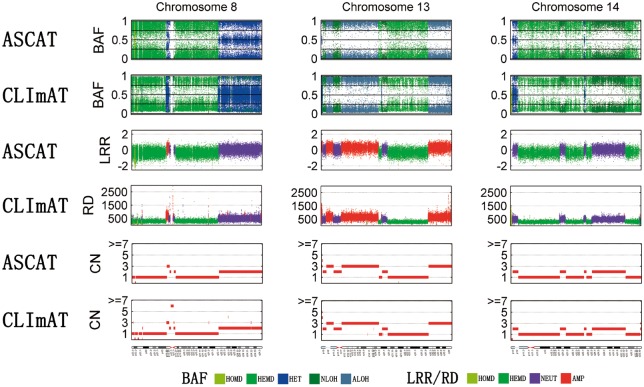
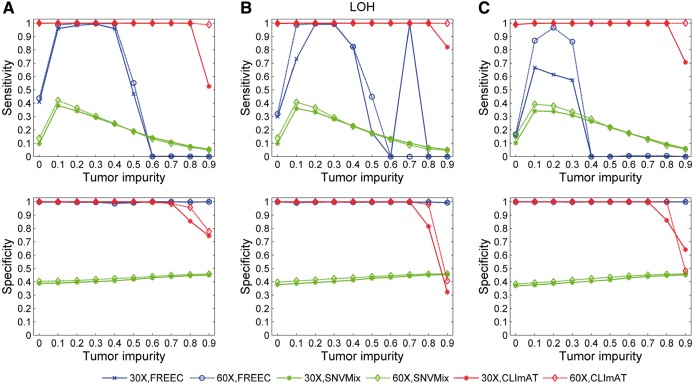
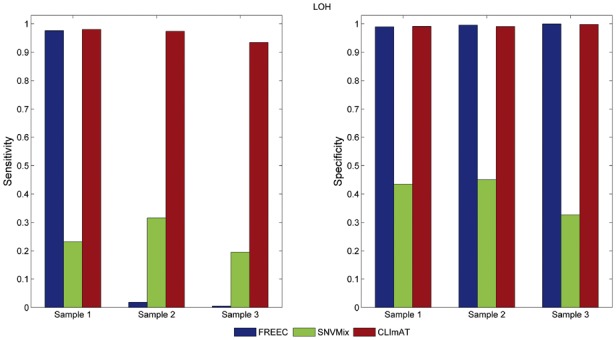
We introduce CLImAT (CNA and LOH Assessment in Impure and Aneuploid Tumors), a bioinformatics tool for identification of genomic aberrations from tumor samples using whole-genome sequencing data. Without requiring a matched normal sample, CLImAT takes integrated analysis of read depth and allelic frequency and provides extensive data processing procedures including GC-content and mappability correction of read depth and quantile normalization of B-allele frequency. CLImAT accurately identifies copy number alteration and loss of heterozygosity even for highly impure tumor samples with aneuploidy. We evaluate CLImAT on both simulated and real DNA sequencing data to demonstrate its ability to infer tumor impurity and ploidy and identify genomic aberrations in complex tumor samples.
The CLImAT software package can be freely downloaded at http://bioinformatics.ustc.edu.cn/CLImAT/.
肿瘤样本的全基因组测序已被证明是一种全面分析癌症基因组中基因组畸变的有效方法。据报道,诸如肿瘤不纯和非整倍体、GC含量和可映射性偏差等关键问题会使复杂肿瘤样本中拷贝数改变和杂合性缺失的识别变得复杂。因此,需要有效的计算方法来解决这些问题。
我们引入了CLImAT(不纯和非整倍体肿瘤中的CNA和LOH评估),这是一种利用全基因组测序数据从肿瘤样本中识别基因组畸变的生物信息学工具。CLImAT无需匹配的正常样本,对读取深度和等位基因频率进行综合分析,并提供广泛的数据处理程序,包括读取深度的GC含量和可映射性校正以及B等位基因频率的分位数归一化。即使对于具有非整倍体的高度不纯肿瘤样本,CLImAT也能准确识别拷贝数改变和杂合性缺失。我们在模拟和真实DNA测序数据上评估了CLImAT,以证明其推断肿瘤不纯和倍性以及识别复杂肿瘤样本中基因组畸变的能力。